
The crystallographic family of products in QUANTA allows the automated addition of solvent (X-SOLVATE), the automatic fitting of ligands (X-LIGAND), de novo density fitting (X-AUTOFIT and X-POWERFIT), general model building (X-BUILD) and Refinement (CNX interface).
These applications provide a complete set of tools, from the tracing of the first map to final refinement and analysis.
The implementation of X-Ray applications within QUANTA provides an integrated environment in which many additional tools from other QUANTA applications, including Protein Design, Protein Health, and Conformational Search, can be used to enhance the model building and refinement processes for proteins and other structures. This chapter describes
This section describes the various X-Ray applications that are integrated within QUANTA. Together, these components provide tools for you to process X-Ray crystallography information from raw data to a refined molecular model of your structure.
Cross references to other chapters in this book and to material in other documentation direct you to detailed information on software applications and functions used to complete the crystallographic process described in the next section.
This section describes the basic activities of the crystallographic process and the software that facilitates each activity.
Use this chapter to determine which components of the Crystallography Workbench Applications you need in your work, as well as when and how to apply them. The remaining chapters of this book describe the crystallographic applications and the CNX interface.
Accelrys' crystallographic software library consists of the following applications that together facilitate the complete structure determination process from crystallographic data:
X-POWERFIT provides a tracing methods for data at resolutions from 1Å to 4 Å. With higher resolution data it is possible to calculate a single Ca trace from a simultaneous analysis of all the bones. At lower resolutions automated interpretation tools streamline the process using secondary structure analysis of the map followed by sequential interpretation using Ca batons. The algorithms available can provide a 500-fold speed up over conventional tracing methods and result in superior quality models.
X-AUTOFIT speeds and enhances the process of fitting coordinates to a SIR, MIR, or MAD map. It skeletonizes electron density maps of proteins, intelligently places alpha carbons to create carbon traces, aligns segments to known molecular sequences, and automatically builds atomic coordinates to fit an electron density map. It can also be powerfully applied in automatically fitting a molecular replacement map.
The X-BUILD application contains many automated model building tools. The application includes interactive regularization, Mote Carlo/grid/gradient real space torsion angle refinement algorithms, and automated water refinement. These and many other tools streamline the process of model building, resulting in a several-fold improvement in productivity.
The integrated X-AUTOFIT and X-BUILD tools include various support applications: a 3D notebook that allows annotation of a molecule, a pointer palette that allows rapid movement around a protein molecule, and validation tools. A Ramachandran plot is always visible while X-BUILD is active, and a Ca plot is always active for the X-AUTOFIT application. Automated and advanced validation is provided specific for crystallographic structure determination as well as full data logging in X-BUILD for data recovery.
CNX is the standard for 3D structure determination of macromolecules using crystallographic or NMR data. The QUANTA interface to CNX export an entire molecular system from QUANTA, including a coordinates file and a script, a principal structure file (PSF), and a parameter file. The interface also launches CNX calculations to perform map generation, simulated annealing, and positional refinement.
You can submit data to CNX interactively or in standalone mode. Results are returned to QUANTA for further manipulation and analysis.
For detailed information about CNX, see Brunger (1992) and the CNX 2006 User Guide (published separately by Accelrys). For more information about the CNX interface, see Setting Up Molecular Systems for CNX.
X-SOLVATE is a rapid method for searching an electron density map for water molecules. The application searches the map for electron density not already filled by atoms and assesses the contacts made with the protein, at all times dealing with symmetry equivalents. Peaks can be assessed interactively and saved as water coordinates. For more information on X-SOLVATE, see Using X-SOLVATE.
The X-LIGAND application allows the rapid searching of regions of significant connected density that may be due to a ligand molecule and not already filled by protein atoms. Different ligands can be fitted to the sorted list of sites, internal degrees of freedom of the ligand can be searched at the rate of thousands per second, and the solution refined using real space torsion angle refinement. For more information on X-LIGAND, see Using X-LIGAND.
The following figure provides an overview of the tasks that the crystallographer can perform to determine and refine a macromolecular structure:
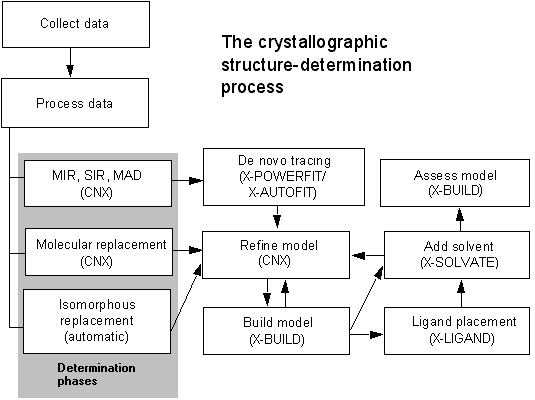
The following paragraphs describe the process outlined in the figure above:
Regardless of where you start in the crystallographic process, you must have a set of structure factors available from. Store these data in a structure factor file (.fob). If no experimental data are available, you can generate a dummy structure factor file. However, in this case, the electron density map that is generated is not physically relevant. For information on how to generate a dummy file.
Experiments performed on an in-house area detector or at a synchrotron facility result in X-Ray data frames that must be processed to extract the intensity of each diffraction peak. X-GEN provides facilities to process these data to the point where a set of merged, corrected intensity measurements are computed and output.
For a new protein structure, phases must be calculated for each diffraction peak, using one or a combination of techniques. Three basic strategies are available for phase determination:
Multiple isomorphous replacement is used when diffraction data are collected for several crystals with various bound heavy metal atoms. You can use the combination of resulting heavy-atom positions to compute phases. Alternatively, you can collect data with multiple wavelength or anomalous dispersion techniques on a single isomorphous derivative to obtain the phase information.
For a protein that is similar to one with a known structure or where a reliable model structure can be generated, you can use the technique of molecular replacement to obtain phases for the new structure. With this technique, a search model of the protein is rotated and translated through the diffraction data to locate orientations that maximize agreement between calculated and experimental data.
Use isomorphous replacement when your protein structure is essentially identical to a known structure. Existing phase data can be used to determine phases from the data for crystals of a mutant or ligand complexed protein.
CNX provides tools for all types of phase determination, including heavy-atom positioning, density modification, and all the methods described above. Molecular replacement is also available within CNX.
When you have obtained phases, you can compute an electron density map and begin the process of building and refining a model for the protein structure. The procedures to be followed for generating a model are somewhat different for each phase determination strategy. However, inspection of the model in the electron density map, manual model building, and refinement are common for all approaches.
If a structure has been solved by MIR, SIR, or MAD, then an initial model for the protein must be constructed from an initial map. X-POWERFIT can automatically trace the Ca atoms form high resolution phased data. For low resolution data, X-POWERFIT allows the identification of secondary structure elements (vectors) and automatic conversion of the vectors to Ca traces. For low resolution structures, as well as the more disordered regions of high resolution structures, X-AUTOFIT can be used to continue the Ca trace construction from skeletonized electron density maps, with built-in intelligence about features of protein structure. This initial Ca trace is converted to an all-atom model using automated tools within the X-AUTOFIT functionality.
The all-atom models generated from X-AUTOFIT or through the process of molecular replacement can be re-built using the X-BUILD application. This allows manual and automated editing of the atom positions and real-space refinement into electron density maps calculated with CNX.
The refinement of model coordinates can be carried out with simulated annealing or traditional least-squares methods within CNX. The QUANTA interface to this program allows automated setup of the required protocols.
It is possible to add ligand molecules automatically to electron densities using the X-LIGAND application. This application includes conformation searching of ligand flexibility and refinement of the ligand to the electron density.
You can use an iterative process for adding solvent and doing small manual rebuilding and careful refinement to produce a final model of the molecule.
You can assess the quality of the final model and also analyze it using the extensive validation facilities designed for detection of crystallographic building error in the X-BUILD application. More general structure analysis tools are found in the Protein Health application in QUANTA and in Insight II.