This chapter describes the various search methods available in the QUANTA Conformational Search application. It is a continuation of information on Conformational Search begun in Chapter 3, Defining Geometric Properties. Read this chapter to get information for planning your conformational search. Then continue with Chapter 5, Performing a Search, for specific procedural information and practice exercises.
Conformational search procedures explore conformational space by varying torsion angles in one or more molecular structures or by varying the relative displacements and orientations of several molecular structures. Therefore, search procedures are accessible only when one or more structures for which a CHARM energy can be calculated are displayed in the viewing areas, and after torsions are defined.
The Conformational Search application provides a variety of search procedures for exploring the conformational space of a molecular system. Search procedures may be broadly classified into two categories:
Systematic (deterministic) search procedures
Stochastic (probabilistic) search procedures
An exhaustive systematic search of the entire conformational space is a very time consuming process for most flexible molecules. The size of the conformational space to be searched increases dramatically with the number of rotatable torsion angles. A thorough search of conformational space can be done in a reasonable length of computer time only for molecules with a small number of rotatable bonds.
There are two factors involved in the increased complexity of a search as the size of a molecule increases:
Both these factors compound to enhance the difficulty of locating minimum energy conformations of a large molecule. A systematic search can be carried out in reasonable length of time only for very small molecules with four or fewer variable torsion angles. In the case of larger systems, either a systematic search is more local, in the sense that the search is carried out only in part of the conformational space, or a stochastic search is used to sample the conformational space.
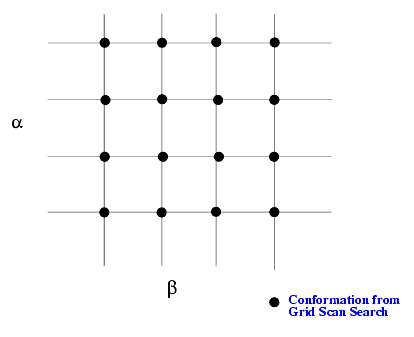
There are two ways to perform a systematic search, Grid Scan and Custom Search. In a Grid Scan search, each specified torsion angle is varied over a grid of equally spaced values. If more than one torsion angle is involved, the variation of the torsion angles are nested. If there are two angles a and b, for a given value of a, angle b assumes a grid of values. If b is the faster torsion angle, the b loop is inside the a loop (see case 1). Although the application is capable of handling up to 10 grid torsions, it is impractical in most cases to employ grid scan for more than four torsion angles.
In a Custom Search, torsion angles are assigned specific values. These values do not need to be equally spaced. This is an advantage in those cases where favorable states of a torsion angle are known from previous modeling studies and the intent is to restrict the systematic search to these values. A further advantage of Custom Search is that it can handle, if so desired, simultaneous changes in several torsion angles. As in Grid Scan, these changes may also be nested.
By definition, stochastic procedures involve random perturbations to a conformation as a method of moving in the conformational space. The nature of the random moves and the criteria employed for accepting the results of the random moves vary from one procedure to another.
In Boltzmann Jump, the torsion angles of a molecule are randomly altered within a specified angular window. In the Peptide Flip procedure, a peptide unit is rotated about the Cai - Cai+1 line. The angle of rotation can be a random value, and the peptide units to be rotated can be chosen randomly from the polypeptide sequence. When there is more than one molecular fragment in the system, the random moves may also involve random reorientation of one fragment with respect to another.
In some stochastic procedures, after each random move a selection criterion is employed to either select or reject the move. The most commonly employed selection criterion is the Metropolis criterion (see citation 1 in the References section):
At higher temperatures, larger upward jumps in energy are likely to be accepted. If the temperature is taken to be zero degrees, no upward jumps in energy are allowed. This special case is employed by the procedure Random Minimization.
The general procedure for performing a conformational search includes the following steps:
1. Define torsions and other geometric quantities.
2. Specify a torsion subset (optional).
3. Select the tag generation method (optional).
7. Provide a filename, supply the tag prefix and number, if prompted.
With these steps completed, the search executes. After the procedure is complete, you have several choices for next steps. You can select:
Conformations generated during a search procedure are saved in binary search files, (filename.csr). Search files contain coordinate data, identifying tags, and optionally, the potential energy for each conformation generated by a search procedure.
The Conformational Search palette, displayed when you select Conformational Search from the Applications menu, includes selections for defining torsions and other geometric properties, selecting a search method, specifying search parameters, and initiating search procedures. The Conformational Search palette also allows you to transfer to the Analysis application without exiting from Conformational Search. Table 14 lists and briefly describes the selections in this palette.
Setup Search in the Conformational Search palette provides access to search procedures and search procedure parameters. When Setup Search is selected, the palette changes by making all the search procedures selectable.
When a search procedure is selected, dialog boxes are displayed to set appropriate parameters for the procedure, then the palette changes to highlight the particular search procedure. All the remaining search procedures are grayed out and not selectable. Do Search becomes selectable.
When Show Search Setup is chosen, a list of parameters for the search procedures that have default parameter settings are displayed in the textport.
A search is executed by selecting Do Search from the Conformational Search palette. A File Librarian dialog box is displayed for you to specify an output search file where the conformations calculated during the search procedure are stored. All search files have the file extension .csr automatically appended to the base filename.
A search procedure can be interrupted by clicking any mouse button. A dialog box offers the option to continue or terminate the search.
All Conformational Search procedures can be run using non-graphical QUANTA by typing the appropriate commands. Set Background Search in the Conformational Search palette facilitates the setup and execution of a non-graphical search. The selection turns on the command record option that automatically records all steps taken to set up and run a search including defining torsions.
When Do Search is selected, the recorded commands are manipulated so that they can be run in a fresh directory. A new directory containing appropriate files is created. In some cases, it may be necessary to manually copy some files. In addition, if a constraints table is to be read and applied during the search, the command record must be edited to include commands to read the table and activate the constraints.
QUANTA is started in non-graphical mode and the recorded search protocol is executed. The current graphical QUANTA session can continue without interference while the non-graphical procedure runs.
Complex search procedures that require a lot of CHARMm processing can generate very large CHARMM.LOG files that may exceed available disc space. To address this issue, select Initialization Options from the CHARMm menu. In the Print Level field, enter an integer smaller than the default value 5 to generate less output for the log file. To suppress nearly all output, enter zero.
Alternatively, the log file can be suppressed altogether by selecting the option, User-specified file, under CHARMm log destination. Use the filename /dev/null.
All conformational search procedures sample conformational space by manipulating specified torsions. In systems comprised of more than one molecular fragment, manipulations of internal coordinates may not be sufficient to explore conformational space.
The selection Rigid Body Manipulation in the Conformational Search palette rotates a structure's coordinates about a randomly oriented vector placed at the center of mass. The extent of rotation is determined by random selection within the specified angular window. After rotation, each fragment is translated along a direction chosen at random by an amount specified by the translation window.
One or more structures may be held rigid during the search. All fragments to be held rigid are loaded into the same MSF. That MSF is designated as the first MSF encountered by the system and all fragments in it are fixed. Remaining fragments held in other MSFs are manipulated during the search procedure.
Tags are unique names that identify conformations. They are assigned during a search procedure just prior to initiating the search. The general format of a tag is:
prefix[code]:number
where prefix is a character string, code designates the search procedure, and number is the rank number (the position in the search file at the time the structure was created by a search procedure). Generally, many conformations are saved into a search file by a single search procedure. The tag format enables the generation of unique tags for each structure saved into the search file.
When a conformation is displayed in either the Conformational Search or Analysis application, the assigned tag is displayed in the upper left corner of the viewing area. Tags also provide a mechanism for identifying the search techniques that generate a set of conformations
When you select Do Search from the Conformational Search palette, a Define Output Search File dialog box is opened with options for defining tag specifications. The application automatically generates a tag containing a prefix, a code, and a starting number for the suffix. The default tag prefix is determined by a set of rules based on the origin of the starting structure at the time the search procedure is started.
The rules for generating the tag prefix are:
A two-letter search key is inserted in the tag if the option Insert Search Key into Tag is selected in the dialog box. Search keys are two-character abbreviations of a search procedure enclosed in brackets. Table 15 lists the keys and the procedures that they identify.
As an example of automatic tag generation, consider a search file generated by the Random Sampling Search method. The fifth conformation in a search file generated by this search procedure will automatically have the tag: filename.msf[rs]:5. If this conformation is then used for a Grid Scan search, the first conformation generated by the Grid Scan search is assigned the tag filename.msf[rs]:5[gs]:1.
The Grid Scan selection in the Conformational Search palette initiates a grid scan search that generates conformations systematically by varying specified torsion angles over a grid of equally spaced values. The number of torsion angles that can be employed in a grid search must be less than or equal to 10. Not more than four torsions are recommended to keep search time reasonable. If more than 10 torsions are defined, a subset of 10 or less must be specified with the Select Torsions tool.
A dialog box is displayed for entering the initial value, a final value, and a torsion increment for each torsion variable. In this box, the numbers entered can be absolute values or values relative to the torsion angles of the starting reference conformation. For example, consider a torsion angle with the initial value of 160o. If the values -30o, 30°, 10o are set in the Grid Scan Torsion Angle dialog box, and if the relative values are specified in the dialog box, the angle will vary from 130o to 190o in steps of 10o. If absolute values are specified in the dialog box, the angle varies from - 30o to 30o in steps of 10o.
A second dialog box is displayed at the start of the grid scan procedure to specify the grid scan processing options. Any combination of these options is selectable. The options are used to specify how the conformations generated on the grid are to be processed. Table 15 lists and briefly describes the options.
The simplest option is just to generate all the conformations and save them into the output .csr file. The CHARMm energy of each generated conformation can also be calculated by turning on the appropriate toggle in the options dialog box. There may be cases where an energy minimization on each conformation is desired. For example, consider a two dimensional grid scan (with two grid torsions a and b) as shown below:
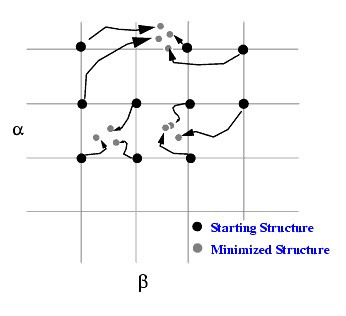
The grid scan explores low and high energy regions. If an energy minimization is not performed at each grid point, the resulting energy variation with a and b is a quick way to identify low and high energy regions. But the conformation will not be a minimum energy structure. Selection of the energy minimization option gives a minimum energy structure but increases the computing time.
The CHARMm energy minimization option is an unconstrained Cartesian minimization process in which all coordinates are adjusted to obtain a minimum energy conformation. During minimization, the grid torsion angles (grid torsion angles a and b in this example) will tend to wander away from the grid point. As shown below, minimizations from some set of grid points may converge towards one region while other grid points may move towards other regions.
Depending on the grid resolution and the size of the grid, this may be a good way to explore low energy regions of this conformational space. A contour plot (see Analysis in the Applications menu) obtained from this search file exhibits a distorted contour shape because the drift of the conformation away from the grid points due to energy minimization is ignored by the contouring scheme which computes a contour of a property varying over a set of grid points.
For this reason, it is often necessary to constrain the grid torsion angles (a and b in the diagram) during the energy minimization using the Constrain Grid Torsions During Minimization option in the dialog box. If this option is selected, during the CHARMm energy minimization, the grid torsion is constrained to the grid point using a penalty energy function. All other torsion angles are allowed to relax in this process. The precision with which the conformation is fixed at the grid point depends on how strained the initial conformation is and how far energy minimization has been carried out. The extent of energy minimization (the number of steps and the convergence criteria) depends on minimization options setup using the CHARMm menu.
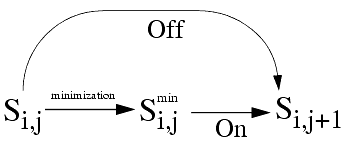
With an energy minimization calculation available for each grid conformation comes another interesting question: how does the program go from one grid point (i,j) to the next grid point (i,j+1) as part of the scan? Consider the scheme shown below:
A conformation, Si,j, is generated at the grid point (i,j). If an energy minimization is performed (with or without torsional constraints), a new conformation ![]() , is obtained. The minimized conformation is then saved into a .csr file. The (i,j+1) conformation can be generated, by suitable torsional changes, from either Si,j or
, is obtained. The minimized conformation is then saved into a .csr file. The (i,j+1) conformation can be generated, by suitable torsional changes, from either Si,j or ![]() . Note that these two starting conformations may differ in bond lengths, bond angles, and in non-grid torsion angles even if the Constrain Grid Torsions option was employed.
. Note that these two starting conformations may differ in bond lengths, bond angles, and in non-grid torsion angles even if the Constrain Grid Torsions option was employed.
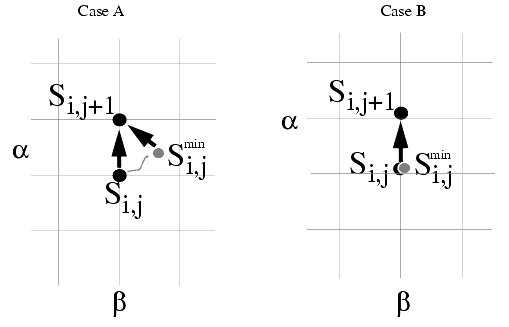
The program provides a choice: to construct Si,j+1 out of Si,j or Si,jmin. This choice is made by either selecting or deselecting the option Keep Minimized Conformation. If this option is not selected, all conformations on the grid are obtained by torsional manipulations of the same initial starting structure. If this option is selected, the conformational changes produced by minimization are carried over to the next state, rather than starting with the original conformation of the structure.
In this case, in scanning through the grid points, adjustments are worked into the structure due to energy minimizations. The resulting structure is used as the reference template from which to generate the subsequent conformations by torsional manipulations.
The following figure illustrates these options:
Display each Structure is an option that enables each structure generated by the grid scan to be displayed as the search procedure progresses. When Display each Structure is not selected the procedure is somewhat faster since the display of the generated structures is suppressed.
The Custom Search method performs a systematic search, varying an arbitrary number of torsion angles to sample a set of torsion angle values. The values assumed by a torsion angle are not required to be grid values at equal intervals.
Custom Search also permits a set of torsion angles to be varied simultaneously. The scope of the search (the torsion angles to be varied and the values to be assigned) is specified in a loop setup file. Prior to selecting Custom Search, a loop setup file is created to describe the scope of the search.
The loop setup file (filename.lsf) is a pre-existing user-defined ASCII file. The file consists of several blocks of data, each block specifying a loop in the search. Even though blocks are listed in sequential order, they specify a nested set of loops in the search. The first block is the outermost loop, and the last block is the innermost loop. Therefore, the torsions specified in the last block will be the fastest varying angles.
Each block can consist of several records (lines). The first record gives the number of torsion angles to vary simultaneously in the loop. The second record gives the angle numbers or names of these torsions. The third and subsequent records list the set of values for these torsion angles. The last set of values is followed by an end of block signal (a record containing the number 999.0). One column is devoted to each torsion angle.
For example, this block of a loop setup file:
assigns six values to torsion angle number 22: -180.0°, -160.0°,
-145.0°, 55.0°, 60.0°, and 65.0°.
In the next example, torsion names are used to assign four backbone conformations to residue number 3:
This example assumes the torsion angles of the residue are defined using a torsion template file that includes entries for phi and psi, and the two torsion angles, phi (3) and psi (3), will vary simultaneously.
A maximum of 10 angles can be included in a given specification, and a maximum of 200 sets of values can be assigned in a block. A maximum of 20 specifications (blocks) can be supplied for a given Custom Search.
Cyclic Modeling is an interactive method that makes it possible to drive the torsion angles of a cyclic molecule. This procedure provides tools for changing the value of any specific torsion angle in the ring system. It then employs CHARMm energy minimization, with suitable dihedral constraints, to achieve low energy structures with the desired torsional values.
Steps involved in initiating Cyclic Modeling are:
1. Select Conformational Search in the Applications menu. Open the Torsions palette by selecting Torsions from the Conformational Search palette.
2. Select Elastic Bond from the Torsions palette and specify the bond to break by clicking on it. The bond will be highlighted momentarily. For cyclic peptides, you can bypass the Pick Torsions and Elastic Bonds steps by selecting Peptide Backbone Torsions instead.
3. Define all torsions using Pick Torsions from the Torsions palette.
4. Select Setup Search and then Cyclic Modeling. The Cyclic Modeling palette is displayed. This palette contains tools to modify the conformation of the molecule. Table 17 lists selections and a brief description of each. Make selections from this palette, exit, and return to the Conformational Search palette.
5. Select Do Search from the Conformational Search palette and specify an output search filename. A new palette is displayed. The conformation of the molecule may be modified by selecting various tools from this palette.
Since cyclic modeling uses CHARMm energy minimization, set up CHARMm minimization options before running Cyclic Modeling. Also minimize the starting structure before you begin.
The Random Sampling search method is a procedure applicable to molecules having a large number of variable torsion angles. Random Sampling randomly changes all defined torsion angles within a predefined angular window. The range of the torsion angle varies during the search procedure and generates new conformations. The search continues for a specified number of steps defined as the Number of Samples. CHARMm energy can be calculated for each randomly altered conformation. CHARMm energy minimization also can be performed. All perturbed conformations are saved to the output search file.
An angular window specification controls the variation in torsion angles for each conformation. If a large angular window value is used, large perturbations are produced. These generate high energy conformations. If a small angular window is used, perturbations are more likely to produce reasonable conformations. However, several cumulative perturbations may be required to significantly migrate from one of part of conformational space to a distant part.
When Random Sampling is selected, a dialog box is displayed to specify Random Sampling parameters. Table 18 lists and briefly describes the options.
Random Minimization performs a minimization based on a random search. No upward jumps in energy are permitted. Torsion angles are used as variables. When Random Minimization is selected from the Conformational Search palette, a dialog box is displayed to set the search parameters. Table 19 lists the options and provides a brief description of each.
When more than one fragment is displayed in the viewing area, the options Rigid Body Translation Window, Rigid Body Angular Window, and Perform Rigid Body Manipulations appear in the dialog box. Perform Rigid Body Manipulations translates and rotates the rigid bodies along a randomly oriented vector placed at the center of mass of each rigid body. The magnitude of the translation and rotation is within the prescribed windows. This option is available only when more than one fragment is displayed in the viewing area.
Conformations displayed in the viewing area are randomly altered within the Torsion Angle Window. If a new conformation is of lower energy than the specification, the next perturbation is performed on that conformation. If the new conformation is of higher energy, it is rejected as a failure, and the previous conformation is retained. The procedure is terminated if a specified number (Maximum Number of Failures) of consecutive conformations results in high energy conformations.
The Boltzmann Jump search method explores conformational space through random perturbation of torsion angles. Boltzmann Jump differs from Random Sampling in that a Metropolis selection criterion is employed to accept or reject perturbed conformations. See citations 1 and 2 in the References section for information on the Metropolis criterion.
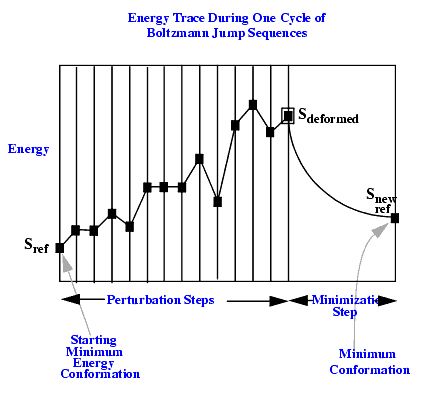
The Boltzmann Jump search is a useful procedure for getting from point A to point B in conformational space using a low energy pathway.
Unlike Random Minimization, in the Boltzmann Jump method, upward jumps in energy are permitted. If an upward jump occurs, a stochastic decision is made to select or reject the new structure.
The probability of selecting the new conformation is equal to the Boltzmann factor defined as e (-DE/RT). The chances of making an upward jump in energy are greater at a higher temperature (T) or at a smaller energy difference (DE).
Starting with an energy-minimized conformation, a random perturbation is carried out within the specified torsion angle window. If the resulting conformation is of lower energy, that conformation is selected. If the perturbed conformation is of higher energy, a stochastic decision is made to select or reject the new conformation.
Random perturbation, followed by a conditional selection of the perturbed conformation, is continued until the root mean square difference (rmsd) in torsion angles between the current conformation and the starting minimum conformation exceeds the specified maximum rmsd. This causes the thermal perturbation sequence to terminate, and a CHARMm energy minimization is performed on the conformation. This procedure is repeated a specified number of times to generate a collection of low energy conformations for the molecule.
A sequence of random perturbation followed by an energy minimization constitute a Boltzmann Jump cycle. An example of an energy profile during a cycle is illustrated in the following figure:
When Boltzmann Jump is selected from the Conformational Search palette, a dialog box is displayed to select parameters for the search procedure. Table 20 lists and briefly describes the options.
There are three parameters that need to be set for a successful execution of the Boltzmann Jump procedure:
The magnitude of the torsion angle window determines the magnitude of the conformational change produced by one perturbation. As this magnitude increases, the chances of producing higher energy conformations increases. At a given temperature, the rate of conformation rejection will increase. If the angular window is decreased, the rate acceptance of conformation by the Metropolis criterion will increase. If too small a value for the window is employed, a large number of perturbations may be required to develop an RMS difference greater then the specified limit.
For a given temperature, it is recommended that trial calculations be performed by gradually increasing the angular window. The highest window value that results in a reasonable number of successful jumps is the optimal value for the window.
The rms limit specified should neither be very small or very large. If it is too small, the perturbation sequence is too short, energy minimization is initiated too soon, and the minimization will most likely bring the conformation back to the starting conformation. If the rms limit is set at too high a value, it may take too many perturbations for the conformation to move very far away from the reference conformation.
Conformation may not change at all if a conformation is unable to develop the specified rms difference. This situation can happen if there is an appreciable energy barrier separating the current class of conformations and the conformations that are farther away. In such a case, a larger rms difference can be obtained only with a higher temperature and possibly a larger angular window. A low value for the rms limit is recommended during trial calculations, followed by iterative increases in the rms limit if the minimization is reached too soon, that is, before a dozen perturbations are performed.
The Hybrid Jump search method is a variation of the Boltzmann Jump. The procedure searches conformational space by varying two sets of torsion variables. The first set, Selection A, is modified using the Boltzmann Jump random perturbation method. The second set, Selection B, uses RIS Sampling.
When Hybrid Jump is selected, the palette changes to accommodate torsion selections for the first set of torsion angles. After you select the first set, the palette changes to accommodate torsion selections for the second set of torsion angles. When the torsion sets are selected, a dialog box displays the parameters for the Hybrid Jump search procedure. Table 21 lists the options and provides a brief description of each.
Do Search in the Conformational Search palette starts the search. The first set of torsion angles (Selection A) is randomly perturbed within the Torsion Angle Window. The second set (Selection B) is assigned to the rotational isomeric low-energy states or user-defined states. This is done for a specified number of conformations (Number of Samples).
The Custom RIS Search method randomly samples torsion angles of a given torsion family from a list of possible states with corresponding statistical weights. The states and the weighting factors must be supplied in a pre-existing, user-defined rotational state file (filename.rsf).
The random search setup file can have up to 20 blocks of data, one block for each torsion family. Each block consists of several records. The first record of each block contains the torsion family name. The second and subsequent records contain specific torsion values and associated statistical weights the family can sample. Up to 20 such records can be supplied, allowing a 20-state model for each family of torsions.
The first record in the .rsf file must contain a 0 in the first space. This record is necessary for other applications using this file though it is ignored by Conformational Search. The last record for each family contains the entries 999.0 and 0.0, to mark the end of each block.
In the following example, five different states are assigned for the families phi and psi, and two states are assigned for the family omega. The statistical weights are automatically normalized to 1.0.
To execute a Custom RIS Search, torsion families must be defined first by reading the appropriate torsion template file (filename.trn). Do Search starts the search procedure.
The RIS Random Sampling search method uses a rotational isomeric state model to limit torsion values to the energetically preferred trans, gauche+, and gauche- states. These states correspond to 180o, -60o, and 60o, respectively.
When RIS Random Sampling is chosen from the Conformational Search palette, a dialog box is displayed to specify the parameters for the search procedure. Table 22 lists the options and provides a brief description of each.
Statistical weights entered in the dialog box are normalized to 1.0, based on their sum. For example, if the values are 10, 4, and 2, the statistical weights will be 0.625, 0.25, and 0.125, respectively. At each perturbation, each torsion angle is reassigned to the trans, gauche+, or gauche- state with the probability of assignment determined by the statistical weights.
The Filter Search method is a variation of the Random Sampling and the RIS Random Sampling procedures. After new conformations are generated, property filters are applied to reduce the number of conformations chosen for further processing. A filter is specified in terms of a geometric property or in terms of an energy value. If the filtering conditions are too stringent, an empty output file may be created. An empty output file is rejected by the Analysis application.
Filter Search offers two methods for generating conformations: a random sampling technique and random choice of a rotational isomeric state (RIS). A dialog box is displayed to choose the generation method.
After the generation method is selected, the Filter Search palette is displayed with additional selections to process conformations, define filters, and save new conformations. Table 23 lists and briefly describes the palette selections.
Filters impose criteria on new conformations. The conformations that meet the criteria proceed to the next step in the process. Filters can apply to torsions, distances (including distances to dummy atoms), energy, radius of gyration, and dipole moment. When Define Filter is selected from the Filter Search palette, the Filter palette is displayed listing property type selections to define the filter. Each filter can include up to 10 different properties.
Properties are combined by selecting an appropriate Boolean operator from the Filter Search palette. The operators .AND. and .OR. are provided for making combinations. These operators appear on the palette after the first property is defined.
Each time a new property is selected, it is combined with the previously defined property by the highlighted operator. For example, let A, B, C, and D be a set of interatomic distances defined using the Geometry palette. If the .OR. tool is highlighted, selection of the Distance property, followed by a distance name specification for each distance, constructs the following filter:
After a filter is defined, two additional selections appear in the palette: .AND. Filter and .OR. Filter. These selections combine filters. For example, two filters are combined by first defining the first filter, selecting .AND. Filter or .OR. Filter, then defining the second filter. For example,
Filter#1 or Filter#2 == (A or B) and (C or D)
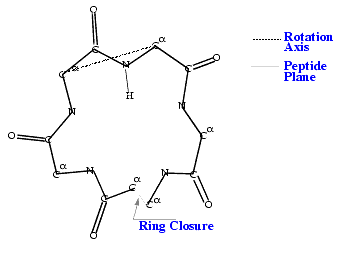
The Peptide Flip search method locates low-energy conformations for cyclic peptides. It is a special random sampling procedure where peptide units are permitted to rotate about pseudobonds connecting alpha carbon atoms. Cyclization algorithms built into the search procedure ensure that a cyclic structure is preserved during torsional manipulation.
In a cyclic system, flexibility of the molecule is often characterized by correlated changes in torsion angles. This is particularly the case where two single bonds are roughly colinear and a crankshaft motion of the intervening section can occur. Such a motion results in a change in the torsion about the first bond and a change of roughly equal magnitude but opposite sign around the other bond.
Such a situation exists approximately for a planar trans peptide unit where a tilt of the peptide plane about the line joining its terminal alpha carbon atoms corresponds to a change in psii. A change of roughly the same magnitude but opposite in sign also occurs in phii+1. As long as the magnitude of these changes is not too large, they produce a local deformation that can be used to rapidly search for better energy minima.
The Peptide Flip procedure utilizes two types of pseudorotations: peptide flips and segment rotations. A peptide flip consists of the rotation of a single peptide unit about the line joining the alpha carbons at both ends of the peptide bond. The following figure locates the rotation axis for a peptide flip:
A segment rotation is a rotation of a sequence of connected peptide units, as a whole, about the line joining the two alpha carbon atoms at the ends of the segment. The following figure locates the axis of rotation for a segment rotation:
Two features of these rotations should be noted:
The Peptide Flip and Segment Rotation procedures affect the conformation of the cyclic system in different ways and therefore, the parameters used for them are different. In most cases peptide flips of 180 degrees produce satisfactory results. On the other hand segment rotations have to be small, preferably between 30 and 60 degrees.
While peptide flips generally do not dramatically alter the alpha carbon atom framework of the peptide after the energy minimization, segment rotation does change the shape of the molecule. Peptide flips do dramatically alter the backbone torsion angles and the hydrogen bonding patterns in the system.
Segment rotation can be viewed as starting a new shape of the molecule, while peptide flips explore the variation of the overall shape. It is for this reason the algorithm employs a series of segment rotations, with each segment rotation being followed by several peptide flips. One starts a new shape for the cyclic system and the other develops energetically favorable peptide plane orientations consistent with that shape.
Results of the Peptide Flip search also depend on:
Peptide flips can be performed on more than one peptide unit. When one or more peptide units are rotated, the peptides that undergo such rotations are selected randomly from the total number of rotatable peptide units. Peptide flips are not performed on a prolyl peptide unit.
Segment rotations can have an angular value that is either fixed or a window for random variation. The length of the segment also must be specified using the number of connected residues that make up the segment to be rotated.
Parameters required for setting up a Peptide Flip procedure are specified in a dialog box. Table 24 lists and briefly describes the options.
For a cyclic peptide with n residues, use a procedure similar to the following set of steps. In this description, each parameter is identified by the name used for it in the dialog box.
1. Choose a segment of length L (Length of the Segment) randomly from n residues. Alternatively, choose a segment of a random size by turning the Use Random Segment Length toggle on in the dialog box.
2. Rotate the chosen segment either through a constant angle or through a random angle within a window of -1/2a to 1/2a. Select Constant Angle for the first option and Angular Window for Random Rotations for the second case.
3. Energy minimize the conformation.
4. Perform M (Number of Peptide Flip Trials before Energy Minimization) trials of peptide flips. Each trial of the peptide flips consists of the following:
5. Recall the lowest energy structure from the M trials done in Step 4. Energy minimize this structure in CHARMm and save the structure. A single execution of Steps 4 and 5 results in one minimized structure being saved into the output structure file.
6. Repeat the Steps 4 and 5 N times (Number of Energy Minimum Conformations per Segment Rotation), so that a total of N structures are saved into the search file.
7. Repeat Steps 1 to 6 a total of K times (Number of Times Rotations are repeated). At the end of this procedure, a total of N¥K energy minimized conformations are saved into the output search file.
Loop Modeling is used to generate low energy conformations in loop regions of a polypeptide or protein. Only the loop portion of the molecule is involved in generating new conformations. The rest of the molecule is held rigid during the procedure. Torsion angles within the loop region are adjusted to ensure acceptable joining of the loop region with the rest of the molecule.
You identify the loop region you want to model by identifying the residue numbers at either end of the loop. An anchor residue is added by the program at each end. These residues belong to the fixed region of the protein.
Torsion angles within the loop are randomly altered. Each torsion angle chi is altered such that each new value is within the chi -0.5 * window and chi +0.5 * window. The window is an angular parameter that is specified in the setup dialog box.
When all torsion angles of the loop are modified, the loop adopts a conformation with anchor residues displaced from their original positions. The loop then goes through rigid and flexible fit procedures and energy minimization. Non-loop portions of the molecule are kept fixed during minimization. Conformation of the protein is saved to an .csr output file. This modeling procedure may be repeated a specified number of times.
The starting and ending residues of the loop are specified along with the torsion windows for the ends and the middle of the loop. Matches may be displayed during the procedure and the loop can be minimized at the end of the design phase.
When Loop Modeling is chosen from the Conformational Search palette, a dialog box is displayed to specify the parameters of the search procedure. Table 24 lists and briefly describes the options.
Conformations can be processed to update pre-existing search or dynamics files without running a search procedure. For example, when a structure does not converge to a real minimum, additional cycles of energy minimization can be carried out.
This chapter describes a variety of conformational search methods available in QUANTA. These methods explore conformational space by varying torsion angles in one or more molecular structures or by varying the relative displacements and orientations of several molecular structures. The search procedures may be broadly classified into two categories:
1. Systematic (deterministic) search procedures
2. Stochastic (probabilistic) search procedures
An exhaustive systematic search of the entire conformational space is a very time consuming process for most flexible molecules. The size of the conformational space to be searched increases dramatically with the number of rotatable torsion angles. In addition, as molecular size increases, the resolution with which each torsion angle needs to be scanned generally also increases. Both these factors compound to enhance the difficulty of locating minimum energy conformations of a large molecule.
A systematic search can be carried out in reasonable length of time only for very small molecules with four or fewer variable torsion angles. In the case of larger systems, either a systematic search is more local, in the sense that the search is carried out only in part of the conformational space, or a stochastic search is used to sample the conformational space.
The general procedure for performing a conformational search includes the following steps:
1. Define torsions and other geometric quantities.
2. Specify a torsion subset (optional).
3. Select the tag generation method (optional).
6. Initiate the search by selecting Do Search.
7. Provide a filename and supply the tag prefix and number, if prompted.
Begin a Conformational Search by selecting Conformational Search from the Applications menu and displaying the Conformational Search palette. This palette includes selections for defining torsions and other geometric properties, selecting a search method, specifying search parameters, and initiating search procedures. Each search is initiated after torsion definition is completed.
1. S. Kirkpatrick, C. D. Gelate Jr., and M. P. Vickie. 1983. Science 220: 671.
2. W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling. 1986. Numerical Recipes. Cambridge University Press.