The most common use of contact maps is to show the inter-residue Ca-Ca distances. Similar properties that are a function of two residues, such as inter-residue van der Waals energy or number of inter-residue hydrogen bonds, can also be plotted in similar fashion. The properties currently displayed as contact maps in this utility are: Ca-Ca distances; Cb-Cb and side chain contact distances; van der Waals interaction energy; electrostatic interaction energy; total interaction energy; hydrogen bonds; and residue type interactions.
Contact Maps in the Protein Design application uses basic x, y plots to analyze the relationship between pairs of residues. Three major categories of contact maps can be calculated: inter-residue distance, inter-residue energy, and residue-residue interaction type. In Protein Design, the contact maps for two proteins can be displayed side by side for easy comparison.
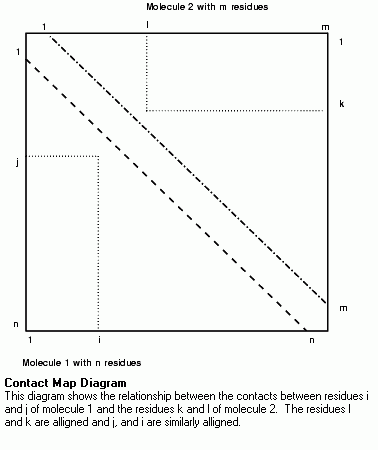
The basic form of the plot for one protein of n residues consists of the x axis, representing residues 1 to n, and the y axis, representing residues n to 1. Therefore, the position (i, j) is color coded according to the value of the property between residues i and j. Since the properties are commutative, the value for (i, j) is the same as for (j, i), then half of the plot is redundant. This allows the top-right of the plot to be used to display similar data for another protein.
The relationship between the sequence alignment and the contact map is shown in the Figures. In the sequence alignment two residues in molecule 1, i and j, are aligned with two residues in molecule 2, k and l. On the contact map, molecule 1 is plotted on the bottom left of the plot and molecule 2 on the top right. The position (i,j) on the bottom left of the plot shows the interaction between residues i and j in molecule 1 and, similarly, the position (k,l) on the top right of the plot shows the interaction between residues k and l. The two positions could be transformed one onto the other by a reflection in the bottom-right to top-left diagonal.
If the Ca-Ca distances of two homologous proteins are displayed side by side, the plot would have a rough mirror symmetry down the diagonal. However, if insertions or deletions are not taken into account in drawing the plot, the two halves of the plot would go out of step resulting in loss of symmetry. Therefore, to make comparison of two side by side plots easier, the axes of each plot incorporate any gaps in the sequence alignment. This means that the plot may have black bands where there are gaps in the protein alignment.
A point on the plot can be picked by double clicking it. The actual residues and contact from that point are reported in the textport. If there are two contact maps on the plot, the same information is reported for the equivalent position on the other map.
These are represented on the contact maps by rectangular boxes that enclose the inter-residue contacts between residues in an alpha helix or beta strand. The sides of the boxes are colored according to the type of secondary structure. Any contacts between residues within the same secondary structure element are close to the diagonal and enclosed in a three-sided box.
The contacts that are shown on a map can also be displayed on the molecule as dashed lines of the same color as the point on the plot. They are located between either the Ca or the Cb atoms of the pair of residues. The amount of information can be undecipherable when displayed on a molecule. It is recommended that the option to select a limited set of residues be used for displaying contact information. The display can also be simplified by having a reduced representation of the protein, such as a Ca trace, and toggling off the visibility of any irrelevant molecules.
Protein sequences should be correctly aligned before using the Difference Contact Map option.
The difference map is calculated by subtracting the contact map for the second molecule (upper right) from the contact map for the first molecule (lower left) with the result being displayed in the bottom left of the plot. The contact map for one of the molecules is shown in the top right of the plot. The exact interpretation of the difference plot depends on which property is being plotted.
The differences maps are colored for increasing magnitude of positive difference using pink (color 14) or red (color 3) and for increasing magnitude of negative difference using pale blue (color 12) and deep blue (color 2).
Three types of distances are mapped: Ca-Ca distance, Cb-Cb distance, and side chain contact distance. These maps are, by default, colored for increasingly close contacts, using white (color 5), pale yellow (color 6), deep yellow (color 4), and red (color 3).
The first two types of maps, Ca-Ca and Cb-Cb, are, by default, scaled to show fairly long range contacts, with all inter-residue contacts less than 16 Å shown. The maps show the overall folding of the structure, and usually have strong diagonal bands that correspond to two close strands in the structure. In this context, a strand might be secondary structure or extended coil. A band in the direction of the leading diagonal, bottom left to top right, correspond to anti-parallel strands, and bands in the direction of the other diagonal correspond to parallel strands.
A difference Ca-Ca distance map can show where there have been gross relative displacement of two strands. Where the contact distances are large (the residues are far apart), are of less interest and can confuse the plot, they can be excluded from the display.
The side chain contact distance map shows the distance between the two closest atoms in the two residues side chains. This map has the same default colors as the Ca-Ca and Cb-Cb plot, but the default scaling is to show short range contacts less than 5 Å. This plot is useful for identifying interacting side chains.
By default, distance difference maps use the absolute difference coloring regime. It is also meaningful to color by the fractional difference, for example, the difference as a fraction of the smaller magnitude of the contact distance for the two molecules. This color scheme is accessed using the Contact Map Options dialog box from the Display Contact Map palette.
Three types of energy maps are available in this utility: van der Waals interaction; electrostatic interaction; total interaction energy. These energy contact maps will identify which residues interact and are colored for increasingly favorable, negative, interaction energies, pale blue (color 12) and deep blue (color 2). For increasingly unfavorable, positive, interaction energies, these maps use pink (color 14) and red (color 3).
The algorithm for the van der Waals energy is a simple 6-12 potential. The atomic radii are taken from the $HYD_LIB/param.par file and depend on the selected atoms being correctly typed. The electrostatic calculation is a simple q(i)*q(j)/r2, with the atomic charges being those assigned to the atom and listed with the atomic information. The total interaction energy is the sum of the van der Waals and the electrostatic interaction energy. The energy calculations can optionally be restricted to the interaction between side chains and, by default, no energy is calculated for residues with the closest atom pair greater than 6Å apart.
The energy difference maps are colored pink or red for increasing positive difference and pale blue and deep blue for increasing negative difference. By default, energy difference maps use the absolute difference coloring regime. As with distance maps (see "Distance Contact Maps" on page 88), the fractional difference coloring scheme can be used.
These two types of maps hydrogen bonds and residue types, are not of quantitative parameters but indicate types of interaction.
Hydrogen bond interactions maps indicate whether H-bond interactions are between mainchain atoms, sidechain atoms, or (if multiple H-bonds exist) within a single atom. The classes of H-bonds and their default colors are:
Residue type interaction indicates where residues have sidechains with atoms less than 6 Å apart. The indication of the type of the pair of residues is:
The following section lists the tools and options found on the Display Contact Maps utility. It is possible to define a range in the sequence table using the Select Active Range tool from the Protein Utilities menu. When this tool is used the calculation is limited to the residues in the active range. This makes the axes range of the contact map limited to those residues.
If you want to compare two structures in side-by-side plots then their sequences must be aligned using the Align and Superpose utility.
This option displays the Contact Map dialog box, that enables you to select the type of contact map. This dialog box contains radio buttons for the different options, plus a toggle for calculating difference contacts.
This option toggles the display of contacts on the first molecule in the viewing area. This corresponds to the contact information in the bottom left of the contact map. By default, this is the first molecule active on the Molecule Management Table. If more than one contact map has been calculated, the most recent one is displayed.
This option toggles the display of contacts on the second molecule in the viewing area. This corresponds to the contact information in the upper right of the contact map. By default, this is the second molecule active on the Molecule Management Table. If more than one contact map has been calculated, the most recent one is displayed.
This option displays the Change Displayed Map dialog box. When two or more contact maps have been calculated, the contacts displayed on the molecule are taken from the most recently calculated map. This dialog box enables you to select an earlier contact map.
This tool limits the interactions between a selected set of residues shown on the contact map or on the molecule display. The residue set must be selected before drawing and calculating a contact map. If the selection is changed while the contacts are displayed on the molecule, then the display is updated to reflect the new selection.
On the Select Atoms palette, one set of residues can be specified for calculating a contact map. Only contacts between the selected residues are displayed, but the axes of the contact map still have all the residues.
This calculates and displays contacts between only the selected residues.
This calculates and displays contacts between the selected residues and the non-selected residues in the same molecule.
This calculates and displays contacts between the selected residues and the rest of the protein including the selected residues.
This tool limits the interactions between selected sets of residues shown on the contact map and on the molecule display. The residue sets must be selected before drawing and calculating a contact map. If the selection is changed while the contacts are displayed on the molecule, then the display is updated to reflect the new selection.
On the Select Atoms palette, two sets of residues can be specified for calculating a contact map. Only contacts between the selected residues are displayed, but the axes of the contact map will still have all the residues.
This option displays the Contact Map Color Ranges dialog box from which the coloring schemes can be changed for the seven coloring regimes.1 Each regime contains a set of ranges that can be edited from Define Coloring Ranges dialog box. For the first five coloring regimes, all distance or energy contacts with values within a given range are colored appropriately for that range.
The Define Coloring Range dialog box shows the maximum for each range and color.2 The user can change the ranges or colors and the number of color bins.
This option displays the Contact Map dialog box from which the default setting can be changed for various contact maps.
This determines if distance or energy difference maps can be colored according to either the absolute value of the difference or the difference as a fraction of whichever is the smaller in magnitude of the values for the two molecules. The default is Absolute Differences.
This determines if the first or second molecule is used to plot the differences between the two molecules on the bottom left of the contact map, and the absolute values for one of the molecules (by default the second molecule) are plotted top right. The default is First Molecule.
The default distance difference cut-off is 6.0 Å. When you look at distance difference maps, the difference in contact distance between residues that are far part is often not of interest and confuses the plot. With this option you can exclude distance differences where the separation distance on both molecules are greater than a given value.
This option sets the energy calculation for side chain only. In addition its use will speeds up the energy contact map calculation.3 The default is 6.0 Å.
This option displays energy or distance contacts for core residues only, such as a fractional solvent accessibility from the default value of 0.5. The default is off.
This option lists the calculated contacts to a file with name MOLECULE_contact_(CA/CB/energy/Hbond).out. This is done as the contact map is calculated. The default is off.
This option highlights in rectangular boxes the areas of the contact map showing interactions between residues in a secondary structure element, such as a alpha helix or beta strand. The default is on.
This tool returns you to the Protein Design palette.