This chapter describes the graphical representation of calculations in the QUANTA Molecular Modeling application. Read this chapter if you plan to use graphical objects to visualize structural data. The objects covered in this chapter include:
In addition, the chapter includes a section on generating and displaying hydrogen bonds.
Graphical objects are displayed in relation to, but separate from, the structures on which they are based. Although each object is a representation of data related to a structure, it is not part of the MSF. However, the option, Attach to MSF in the Object Creation dialog box is checked as a default selection. This option allows you to attach an object to an MSF so that it may be moved and deleted with the structure. Using the Attached column of the Object Management Table, you may toggle the object/MSF attachment. The Object Management Table also provides object display, naming, and deletion options. Each graphical object is uniquely identified so that you can manage it throughout a QUANTA session.
Some objects such as ribbons, Connolly solvent surfaces, and grid contour meshes are generated within QUANTA. Other objects such as brick maps and 3D vectors are generated from results of calculations outside of QUANTA.
Ribbons are graphical objects that trace peptides, protein backbones, or any other molecular chain through guide atoms that occur in all residues of a molecular structure. The ribbon-drawing algorithm used in QUANTA is from methods cited in references 1 and 2 in the Reference section of this chapter.
In QUANTA, the alpha carbon and carbonyl oxygen atoms of each protein residue act as default guide atoms for drawing a ribbon. Guide atoms occur in all residues. The ribbon is offset 1.2 Å from the alpha carbon position. Ribbon defaults are set to optimally display alpha helices in proteins.
Guide atoms, ribbon width, number of threads, and ribbon color can be changed through the Draw menu by accessing the pull-right menu Object Parameters and selecting Ribbon Parameters. A dialog box enables you can make changes.
Complete the next exercise to become familiar with the procedure for generating and manipulating a ribbon. To complete this exercise, you need to use the MSF mypeptide.msf. This file was created as part of an exercise in the section Creating a Peptide in Chapter 4. Complete that exercise before you proceed.
Display the File menu and select Open. In the Open MSF File Librarian dialog box, select mypeptide.msf from the scrolling list.
Click the Replace button and then the Open button. The structure contained in the file mypeptide.msf is displayed in the viewing area.
Display the Draw menu and select Create Objects. A dialog box offers options for creating several graphical objects.
Select Ribbon Structure and then enter ribbon and 8 in the respective Object Name and Object Color data entry fields.
Accept the default option, Attach to MSF. The name of the open MSF, mypeptide.msf, is displayed in the box next to this selection. The ribbon is attached to this MSF and can be moved and deleted with the MSF.
The dialog box is closed, and a ribbon consisting of six strands is drawn through the Ca and O atoms in the backbone of mypeptide.msf.
The four middle strands are displayed using color 8. The two outside threads are a different color, providing a 3D representation of the backbone chain.
The Object Management Table opens over the Molecule Management Table, displaying the name and status of the ribbon.
Grid contour meshes are graphical objects that provide an efficient and visually effective method of displaying surfaces on atoms. All active atoms or a selection of atoms may be used in generating the grid. Atom surfaces are determined by the van der Waals radii of the current atom selection. Selected atoms are defined in the context of other atoms in the structure.
When a grid contour mesh is created about a methyl group attached to a phenyl ring, you may include or exclude the atoms in the phenyl ring. Including the atoms in the ring creates a grid that is open at the point where the methyl group is attached to the phenyl ring. If the atoms in the ring are excluded, an enclosed grid is created.
1. Create a grid contour mesh for all atoms.
Display the Draw menu and select Create Objects. In the dialog box for creating objects, select the option Grid Contour Mesh. Then enter grid1 and 4 in the Object Name and Default Object Color data entry fields, respectively.
Select the option Attach to MSF and the name of the open MSF, mypeptide.msf, is displayed in the box next to the option.
Click the OK button and a dialog box offers options for the selecting atoms to define the grid.
Accept the default text all by clicking the OK button. A dialog box appears with options that select atoms to define the context of the grid.
Accept the default text all by clicking the OK button The dialog box is cleared from the window, and a grid contour mesh is displayed around the structure mypeptide.msf in color 4.
Two graphical objects are now displayed with the structure mypeptide.msf: the ribbon and the grid contour mesh. The Object Management Table lists both objects.
Turn off the display of both objects by selecting the top cell in the Displayed column. Both objects are removed from the screen, and the entry for each in the Displayed column of the Object Management Table changes from yes to no.
2. Create a Grid Contour Mesh from Selected Atoms.
Display the Draw menu and select Create Objects. A dialog box offers options for creating several graphical objects.
Select the option Grid Contour Mesh, then enter grid2 and 14 in the Object Name and Object Color data entry fields, respectively.
Select Attach to MSF and the name of the open MSF, mypeptide.msf, is displayed next to the option.
Click the Interactive button and the Atom Selection Utilities and Select Atoms palettes are displayed.
From the Select Atoms palette, select Include and Pick Atom.
In the viewing area, pick any three atoms in different areas of mypeptide.msf, and the ID for each selected atom is displayed. The three picked atoms define the center of each portion of the grid contour mesh.
From the Select Atoms palette, select Finish and the palettes are removed from the screen. A dialog box offers options for selecting atoms to define the context of the grid.
Enter the text All and click the OK button. The grid contour meshes are displayed around the selected atoms in the viewing area and the Object Management Table lists the object GRID2 and its status.
If your structure has 3D vector information, you can display a vector for each atom. The information is stored as fourth parameter information of type REA3 in the .msf.
Vector data representing forces can be transferred from CHARMm to the .msf using Get Fourth Parameter in the CHARMm menu and selecting Forces from the pull-right menu. This extra information is labeled FORCE.
Vector data from external sources can be imported into a .msf by selecting Incorporate Data from the Edit menu, selecting Vectors from the pull-right menu, and specifying a file from the list in the File Librarian that is displayed.
1. Select Create Objects from the Draw menu, select 3D Vectors in the dialog box that is displayed, then click OK. A dialog box is displayed prompting you to enter the label under which the vector data is stored. A list of labels and their types is output in the textport.
2. Enter a label of type REA3, then click OK. A new dialog box is displayed.
3. Select the options you want to use for your vector display. The linear scaling factor may need trial-and-error adjustment depending on the magnitude of the vectors to be represented. By default, the color of a vector is related to its magnitude and the colors are scaled from number 7 to 14.
4. When you have selected the options you want in this dialog box, click OK. Vectors are displayed as arrows attached to atoms on your structure.
You can generate a QUANTA plot file (.qpt) to move graphical information to or from another application, to generate hardcopy images, or for other purposes. When a .qpt file is available, you can display the graphical information in it by creating a QUANTA plot file object. This display mechanism is an alternative to using Plot Molecules/Preview in the File menu.
1. Select Create Objects from the Draw menu, choose QUANTA Plot File in the dialog box that is displayed, then click OK. A File Librarian is displayed.
2. Select the .qpt file that you want to display, then click OK. A dialog box is displayed.
3. Select one of the two display options for the extent of display that you want, then click OK. The 2D image is displayed in the model window. You may have to scale this window to see a complete view of the structure.
In the lower left corner of the molecule window, you can display a scale that shows the current color values. You also may add a maximum and minimum value to this display to indicate the value range to which these colors apply. Typically, this scale is used in conjunction with work where you have colored the structure according to some property, such as atomic charge.
1. Select Create Objects from the Draw menu, choose Color Scale in the dialog box that is displayed, then click OK. A dialog box is displayed.
2. In the dialog box, indicate the range of color numbers, toggle Annotate Scale on or off and, if you are annotating the scale, supply minimum and maximum values, then click OK. A color scale is displayed in the molecule window.
The Object Management Table is the primary means for managing objects. Each graphical object is uniquely identified so that you can manage it throughout a QUANTA session. The Object Management Table lists:
Each object is manipulated with a set of toggles that are activated by selecting the appropriate cell in the Object Management Table. The table functions in a manner similar to the Molecule Management Table. For example, you can mask the display of grid1 generated in a previous section by selecting the cell in the Displayed column of the Object Management Table that corresponds to grid1. When you make this selection, the cell information changes from yes to no and the grid is removed from the viewing area.
Complete this exercise to become familiar with the Object Management Table.
The Object Management Table lists three objects: RIBBON, GRID1, and GRID2.
Select the Displayed cells for RIBBON and GRID1. All three objects are displayed, and each Displayed cell reads yes.
2. Mask the display of one object.
Select the Displayed cell again for grid1. The cell information changes from yes to no and grid1 is removed from the viewing area. The ribbon and grid2 remain displayed.
3. Detach an object from its associated MSF.
Select the Attached cell for grid2. The cell information changes from yes to no. No change is observed in the viewing area.
Go to the File menu and select Open. In the Open MSF File Librarian dialog box, select PEPTIDE.msf from the scrolling list. Then select the Replace option and click the Open button. The mypeptide structure is replaced by the PEPTIDE structure and the RIBBON and GRID1 objects are deleted. Grid2 remains displayed and listed in the Object Management Table.
4. Attach an object to an MSF.
Select the Attached cell for GRID2. The cell information changes from no to yes, and the file name PEPTIDE.msf appears in the MSF column.
5. Change the name of the object.
Select the Name cell for GRID2 and a dialog box offers the option to rename the object.
Enter TESTGRID in the name field and click the OK button. The name in the Object Management Table changes from GRID2 to TESTGRID.
6. Close the MSF and delete the remaining object.
Go to the File menu and select Close. When the dialog box opens, select Close All then OK. The peptide structure is removed from the viewing area and the object is deleted
Connolly solvent surfaces are graphical objects that are used to study the shape, depth, and chemical characteristics of receptor sites in a molecular structure. These surfaces provide effective representations of such important atomic and molecular properties as surface-solvent accessibility and electrostatic potential.
In the Calculate menu, the Solvent Surfaces selection provides an interface to an external program that calculates Connolly surfaces by rolling a probe sphere over the surface of a molecular structure. See citation 3 in the References section of this chapter.
Calculations can be run on hosts other than the local system. To do this, display the Calculate menu and select Select Host. In the dialog box that opens, select a host. Then proceed with the calculation by returning to the Calculate menu and selecting Solvent Surfaces.
Calculation results are written to a surface file, filename.srf, and a solvent surface is displayed as a graphical object. You can have the map be displayed automatically on completing the calculation or at a later time by selecting Create Objects from the Draw menu.
Complete the following exercise to become familiar with the process of creating a Connolly surface. The exercise uses the structure, nitrobenzene created in the Crystal Modeling section of Chapter 5. If you have not completed the first part of the exercise in this chapter, do so before you proceed.
Open nitrobenzene.msf and display the structure in the viewing area.
2. Start a Connolly solvent surface calculation.
Display the Calculate menu and select Solvent Surfaces. From the pull-right menu that opens, select Calculate. A dialog box provides surface options. See Table 29 for a list of the options and a brief description of each.
Wait for Calculation to Complete
Probe Radius: 1.4
Dot Density: 20.0
Solvent Accessible Surface
Use Active Atoms
These selections specify the type and display of the surface and the atomic context for the calculation.
Click the OK button and the Select Atoms and Selection Utilities palettes open.
From the Select Atoms palette, select Include and All Atoms, and then select Finish.
In a File Librarian dialog box, enter the name:
Click the Save button and the filename extension .srf is automatically entered in the data entry field. The calculation starts and the cursor changes to a watch while the calculation is in process.
4. Display the Connolly solvent surface as a graphical object.
When the calculation of the surface is complete, a dialog box offers the opportunity to enter a name for the surface object.
Click the OK button and a dialog box opens with options for selecting the atoms to be surfaced and the color of the surface.
All Atoms
Use Atom Colors
Display Big Dots
Click the OK button and the surface is displayed over all atoms, using the atom colors. The Object Management Table lists Connolly1 and its status.
In the Object Management Table, toggle the Delete cell of the Connolly1 object listing from no to yes. The surface is deleted from the viewing area. However, the file nitrobenzene.srf is not deleted.
6. Re-create the surface object.
Once a surface file has been created, the surface object can be displayed at any time.
Display the Draw menu and select Create Objects. In the dialog box that opens, select:
Solvent Surface Maps
Attach to MSF
Accept the default color and click the OK button. In the File Librarian dialog box that opens, select nitrobenzene.srf from the scrolling list and then click the Open button.
A dialog box opens for entering information on a variety of object display parameters. See Table 30 for a list of options and a brief description of each.
All Atoms
Single Color
Display Plus Symbols
Click the OK button. The surface is displayed and the Object Management Table opens listing the Connolly2 object.
7. Delete the graphical object.
Repeat the process used for Connolly1.
In the Object Management Table, toggle the Delete cell of the Connolly2 object listing from no to yes. The surface is deleted from the viewing area. However, the file nitrobenzene.srf is not deleted.
Probe Interaction calculates empirical energy field maps, called brick maps, and displays them. A probe is used to sample the environment of the displayed structure. The probe is a point charge with van der Waals characteristics. As the structure is sampled, the energy is calculated between the probe and the structure at all points within a sampling box.
The energy calculated is the van der Waals energy, the electrostatic energy, or the sum of both energies. The van der Waals energy is calculated using the atom types currently assigned. The electrostatic energy is calculated using the charges currently assigned to the atoms.
The results of the calculation are written into a specified file, filename.mbk, and a brick map is displayed as a graphical object. The number of contours contained in this map and the color of each contour can be changed within QUANTA. The map can be displayed automatically on completion of the calculation, or at a later time.
In this section, empirical energy field maps are calculated for the interaction of a proton with nitrobenzene (nitrobenzene.msf). Procedures include:
Complete the following exercise to become familiar with the procedure for generating and manipulating a brick map.
1. Start a Probe Interaction calculation.
Display the Calculate menu and select Interaction Maps. From the pull-right menu that opens, select Probe. A dialog box offers probe interaction options. See Table 31 for a list of the options and a brief description of each.
Wait for Calculation to Complete
van der Waals + Electrostatics
Probe Name: prot (four letters only)
Probe Charge: 1.0
Box Dimensions (angstroms): 10
Grid Size (angstroms): 0.5
Maximum Energy: 100
Minimum Energy: -100
Click the OK button and the message line reads:
Pick one atom - Pick any palette tool to quit.
Pick atom for center of box.
Pick the nitrogen atom in the structure. A File Librarian dialog box requests a name for the brick map file (.mbk). In the text entry field, enter the text:
Click the Save button. The calculation starts and the cursor arrow changes to a wait watch.
2. Display the brick map as a graphical object.
When the calculation of the brick map is completed, a dialog box requests a name for the map graphical object. Enter the name:
Click the OK button. A dialog box offers options for selecting the area of the brick map to be displayed.
Click the OK button. A dialog box asks for the number of contour levels to map.
Click the OK button. A dialog box offers options for specifying the values and colors for each contour level.
1st contour level: -4.0
2nd contour level: 2.0
1st contour color: 14
2nd contour color: 13
Click the OK button and the brick map is displayed using the new contour values and color specifications. The energy value at grid intersections in each contour is displayed in the grid's associated color in the lower-right corner of the viewing area. You may wish to scale down the display using the Dial Emulator to see the entire surface.
The brick map object can also be created by selecting Create Objects from the Draw menu.
3. Delete the brick map object.
In the Object Management Table, toggle the Delete cell of the probemap1 object from no to yes. The surface is deleted from the viewing area, but the file nitrobenzene.mbk is not deleted. The brick map is deleted and no longer displayed.
The Molecule Interaction function computes the interaction between two molecules: a probe and a target. The interaction energy between probe and target is the sum of two nonbond interactions:
The probe molecule is tumbled about the target at specified angles and increments. An energy calculation is performed at each point of a grid superimposed on the target, using all possible combinations of probe rotation angles. A contour map is computed by taking the minimum energy value from each grid point. For fine grids or small rotation increments, the molecule interaction calculation can be lengthy. For example, for a probe tumble angle of 60°, 216 energy calculations are performed at each grid point.
The electrostatic and van der Waals interactions of a probe molecule, water, and target molecule, search1.msf, are calculated in the following exercise and displayed as brick maps. The structure, search1, was constructed in Chapter 2. If you did not complete the exercises in Chapter 2, do so before proceeding with this exercise.
1. Build a water molecule probe.
Display the Applications menu, select Builders, and then select 2D Sketcher from the pull-right menu. In the ChemNote window that opens, place an O (oxygen) atom in the viewing area. The 2D function automatically adds the appropriate hydrogens.
Display the ChemNote File menu and select the Return to Molecular Modeling function. A dialog box asks if the changes should be saved.
Click the Yes button. In the File Librarian dialog box that opens, enter the text:
Click the Save button. The ChemNote window is removed from the screen and a dialog box offers options for displaying MSFs.
Use the new molecule water.msf only
Click the OK button and the structure stored in water.msf is displayed.
2. Open the target molecule file (search1.msf).
Display the File menu and select Open. In the Open MSF File Librarian dialog box, select the file search1.msf from the scrolling list.
Click the Open button and both the water molecule and the search1 molecule are displayed in the viewing area.
Select Reset View from the Geometry palette to reposition the MSFs in the center of the viewing area.
3. Set up the molecule interaction calculation.
Display the Calculate menu and select Interaction Maps. From the pull-right menu that opens, select Molecule and the Molecular Interaction Grid dialog box opens. See Table 32 for a list of the options and a brief description of each.
Wait for Calculation to Complete
van der Waals + Electrostatic
Probe Tumble Increment:180
Box Dimensions (Angstroms): 15
Grid Size (Angstroms): 1
Maximum Energy: 100
Minimum Energy: -100
Click the OK button. The 180° increment and 1 Å grid size expedite the calculation. Values in the vicinity of 60° and 0.1 Å are more typically used in accurate molecule interaction calculations.
4. Define the atom to be the center of the box.
A message in the message line reads:
Pick an atom - Pick any palette tool to quit. Pick an atom for center of box.
Pick the search1.msf ring carbon that is the connection point for the longest branch. In the File Librarian dialog box that opens, enter the name of the brick map.
5. Select the probe molecule and begin calculation.
In the dialog box that asks for which molecule to display, select the structure:
Click the OK button. The calculation begins and the cursor changes to a watch until the calculation finishes in approximately 10 minutes, depending on the hardware. When the calculation is complete, a dialog box is asks you to choose a save option.
Create New Generation of water.msf
The structure is saved and the textport reads:
The old structure is renamed water.msf,001
A dialog box asks for the name of the map just calculated. Enter the name:
Click the OK button. A dialog box asks which molecule to attach the interaction map to.
Select search1.msf from the scrolling list.
Click the OK button. A dialog box offers options for selecting the area of the brick map to be displayed.
Click the OK button. A dialog box asks for the number of contour levels to map.
Click the OK button. A dialog box offers options for specifying the values and colors for each contour level.
1st contour level: -3
2nd contour level: 3
1st contour color: 7
2nd contour color: 8
Calculation time depends on the hardware, but is usually short. When the calculation is complete, a brick map of the contours is displayed. The low-energy attractive regions of the surface are displayed in color 7, default green. High-energy repulsive regions are displayed in color 8, default purple.
Rotate and translate the structure to observe the relationships between chemical structure and contour shape.
7. Delete the brick map object.
In the Object Management Table, toggle the Delete cell of the mol_inter object listing from no to yes. The surface is deleted from the viewing area, but the file mol_inter.mbk is not deleted.
Brick maps and other graphic objects are not saved when QUANTA is exited. However, graphic objects, a brick map in this case, can be reconstructed without repeating the calculation.
Display the Draw menu and select Create Objects. In the Graphic Object Generation Options dialog box that opens, select the option:
Object Name: probemap1
Object Color (1-14): 1
Select search1.msf from the scrolling list.
Click the OK button. From the scrolling list in the File Librarian dialog box, select mol_inter.mbk, and then click the Open button.
In the new dialog box that is opens, select the option:
Click the OK button. In the contour level dialog box that opens, enter the value:
Click the OK button. In a dialog box for entering the contour levels and color numbers, note the minimum and maximum values reported, and enter the values:
1st Contour: -3
2nd Contour: 3
1st Contour Color: 7
2nd Contour Color: 8
Click the OK button and a brick map of the contours is displayed. The low-energy attractive regions of the surface are displayed in color 7, default green. High-energy repulsive regions are displayed in color 8, default purple.
Rotate and translate the structure to observe the relationships between chemical structure and contour shape.
9. Delete the brick map object.
In the Object Management Table, toggle the Delete cell of the mol_inter object listing from no to yes. The surface is deleted from the viewing area, but the file mol_inter.mbk is not deleted.
You can calculate and generate a brick map display based on electrostatic interactions, ignoring van der Waals interactions.
Complete the following exercise to show the interaction of a water molecule with search1.msf, without van der Waals interactions being modeled in the calculation.
1. Start the molecule interaction calculation.
Display the Calculate menu and select Interaction Maps. Then select Molecule from the pull-right menu that opens and the Molecular Interaction Grid dialog box opens. See Table 32 for a list of the options and a brief description of each.
Wait for Calculation to Complete
Electrostatic Calculation
Probe Tumble Increment:180
Box Dimensions (Angstroms): 10
Grid Size (Angstroms): 1
Maximum Energy: 100
Minimum Energy: -100
Click the OK button and the 180° increment 1 Å grid size expedite the calculation. Values in the vicinity of 60° and 0.1 Å are more typically used in accurate molecule interaction calculations.
A message in the message line reads:
Pick any atom - Pick any palette tool to quit.
Pick an atom for center of box.
Using the mouse, select the same ring carbon used in the previous calculation.
A File Librarian dialog box asks for the name for the map file. Enter the filename:
Click the Save button and a dialog box asks which molecule is the probe. From the scrolling list, select:
Click the OK button. The calculation begins and the cursor changes to a watch. When the calculation is complete, a dialog box asks you to choose a save option.
Create New Generation of water.msf
The structure is saved and the textport reads:
The old structure is renamed water.msf,002
2. Display the brick map as a graphical object.
When the calculation of the brick map is completed, a dialog box requests a name for the map graphical object.
Click the OK button and a dialog box opens for attaching the graphical object to an MSF. Select the scrolling-list entry:
Click the OK button. A dialog box offers options for selecting which area of the brick map to display.
Click the OK button and a dialog box asks for the number of contour levels to map.
Click the OK button and a dialog box offers options for specifying the values and colors for each contour level.
Click the OK button. The brick map is displayed with electrostatic interactions in contours of light blue.
3. Delete the brick map object.
In the Object Management Table, toggle the Delete cell of the electro1 object listing from no to yes. The surface is deleted from the viewing area, but the file electro1.mbk is not deleted.
The Maps Management table provides an interface to the general-purpose volume visualization capabilities of QUANTA. 3D volume information is stored in brick maps, which are used for storing electron density, molecular probe maps, volumes, electrostatic potential, and molecular surface information generated within QUANTA. In addition, the maps can be generated by external programs for display within QUANTA. The molecular or volume properties that these maps contain are displayed within QUANTA as chicken-wire contours or solid surfaces.
The Maps Management table is available in any application in QUANTA and provides a spreadsheet-like interface to tools that manage and display the 3D information held in QUANTA brick map files. Its display can be turned on or off using the Map Table pull-right menu on the Draw menu. These tools convert map formats to QUANTA-compatible brick maps and define active maps, contour levels and display styles. Additional tools save and retrieve the contents of brick map display lists and purge the brick map database of undisplayed bricks to reduce memory requirements.
The following figure shows the Maps Management table. The table has the following components:
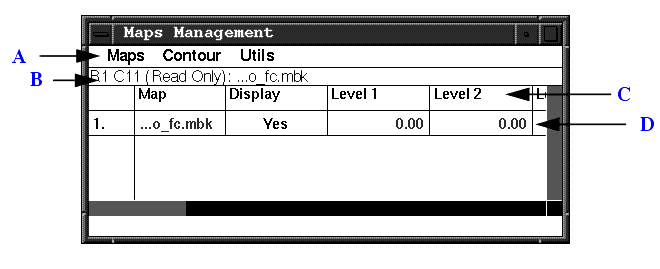
Some tools available on the Maps Management table are not relevant within X-LIGAND, X-SOLVATE, X-AUTOFIT, X-POWERFIT, and X-BUILD. These applications automatically control the map display; changes to some general map options can interfere with the tools' use of the maps, with serious detriment to speed and results. Please refer to the X-Ray Structure Analysis User's Guide for further details if you use these applications.
The Electrostatics selection in the Calculate menu provides the ability to calculate and display an electrostatic potential energy surface on a molecule in either:
The electrostatic potential can be calculated using partial atomic charges obtained from a previous CHARMm calculation or with the VSS method based on the work of Giessner-Prettre and Pullman. (See Method II described in citation 6 in the References section of this chapter.) 3D potentials can be displayed as a grid map or a dot matrix.
On the electrostatic potential grid map, large negative potential contours correspond to very electronegative regions on the van der Waals surface and appear where negative partial atomic charges control the electrostatic potential. Regions corresponding to positive electrostatic potential occur where the resulting charge is positive. A color-coded scale of electrostatic potential is listed in the lower-right corner of the viewing area. The color display scale can be adjusted to fit the data for the displayed structure.
A dot matrix display uses dots to outline the van der Waals surface. Color may also be used to display areas of relative electronegativity or positivity.
Complete the following exercise to build the structure cyclohexanol and then generate an electrostatic grid map for it:
1. Create cyclohexanol using ChemNote.
Display the Applications menu, select Builders, and then select 2D Sketcher from the pull-right menu that opens. In the ChemNote window, sketch the structure cyclohexanol, using appropriate bond, ring, and atom selections. If you have never used ChemNote, complete the exercises in Chapter 1 to become familiar with its operations.
2. Return the cyclohexanol structure to Molecular Modeling.
Display the ChemNote File menu and select Return to Molecular Modeling. A dialog box asks if the changes should be saved.
Click the Yes button. In the File Librarian dialog box that opens, enter the text:
Click the Save button. The ChemNote window is removed from the screen, and a dialog box offers options for displayed MSFs.
Use the new molecule cyclohexanol.msf only
Click the OK button and the structure is displayed in the viewing area.
Rotate the structure to visualize the orientation of the hydroxyl hydrogen relative to the ring atoms.
3. Move the hydroxyl hydrogen.
From the Modeling palette, select Torsions. The Select Torsions palette is displayed and the message line reads:
Pick 1st atom defining torsions or pick FINISH to continue
Pick the carbon atom attached to the oxygen and the message line reads:
Pick 2nd atom defining torsions or pick FINISH to continue
Pick the oxygen atom as the second atom involved in the torsional rotation and the message line reads:
Pick 3rd atom defining torsions or pick FINISH to continue
From the Select Torsions palette, select Finish.
Dial Set 3 is displayed in the Dial Emulator. Use the Torsion 1 dial to rotate the hydroxyl hydrogen to a position in which it is trans (180°) to the hydrogen on the carbon that is attached to the hydroxyl oxygen. Ignore the numbers in the lower left of the viewing area.
From the Modeling palette, select Save Changes. A dialog box offers options to save the new coordinates of cyclohexanol.msf.
Click the OK button and the coordinates are saved.
Display the CHARMm menu and select Minimization Options. A dialog box offers minimization setup options.
Number of Minimization Steps: 15
Coordinate Update Frequency: 1
Energy Gradient Tolerance: 0.000001
Energy Value Tolerance: 0
Initial Setup Size: 0.02
Step Value Tolerance: 0
Click the OK button and the new minimization parameters are established.
From the Modeling palette, select CHARMm Minimization. The CHARMm calculation is begun and stops when the rms force is less than 0.000001.
6. Save the minimization changes.
From the Modeling palette and select Save Changes. A dialog box offers the option to save the new coordinates of cyclohexanol.msf.
Click the OK button and the minimized coordinates are saved.
7. Calculate electrostatic potential.
Display the Calculate menu and select Electrostatic. From the pull- right menu, select Potential and the Calculate Electrostatic Potential palette is displayed.
Select Calculate 3D Surface, Calculate from Partial Charges, and 3D Surface Parameters.
From the Setup 3-D Surface Parameters dialog box, select the default options:
Reentrant
Solvent Probe Radius: 1.4
Medium
Big Dots
Grid Contour Mesh
8. Run electrostatic potential.
From the Calculate Electrostatic Potential palette, select Run and Wait. In the File Librarian dialog box that opens, enter the text:
Click the Save button. The electrostatics calculation starts and the cursor changes to a watch. You can follow the calculation status in the textport. When the calculation is completed, the cursor returns to an arrow.
From the Calculate Electrostatic Potential palette, select Display Results. The Display Potentials palette and a File Librarian dialog box are displayed.
From the File Librarian, select the 3dcyclo.epo file and then click the Open button.
From the Display Potentials palette, select Show 1-Molecule Potential and a dialog box opens for entering an object label.
and click the OK button. A grid map appears in colors described in the color scale in the lower-right corner of the viewing area. The colors indicate the potential near the surface of the molecule.
The Object Management Table opens listing the object, potential1, and its status.
In the Display Potentials palette, select Set Contour Levels and a dialog box opens with a set of eight Potential Value data entry fields. The minimum and maximum potential values (in units of kcal/mol) for the simulation are reported at the top of the dialog box. These values can be used to create more realistic contour values.
Referring to the values at the top of the box, enter the value for the Minimum Value of Potential in the Potential Value 1 data entry field.
Enter the value for the Maximum Value of Potential in the Potential Value 8 data entry field. These are the upper and lower limits for the potentials of the electrostatic field for cyclohexanol in this calculation.
Click the Auto Scale button. The values in the Potential Value 2 through Potential Value 7 boxes are automatically incremented between the minimum and maximum potential values that you enter. You can also set values 2 through 7 manually.
Click the OK button to display the contours using the new values.
From the Display Potentials palette, select Finish. The Display Potentials palette is closed and the Calculate Electrostatic Potentials palette is redisplayed.
The electrostatic potential can also be displayed as a dot surface. The dot surface electrostatic potential is calculated and displayed in the same manner as the electrostatic potential grid map.
Complete the following exercise to display a dot surface electrostatic potential for cyclohexanol.
1. Set up a dot surface electrostatic potential.
Display the Calculate Electrostatic Potentials palette.
Select Calculate 3D Surface, Calculate from Partial Charges, and 3D Surface Parameters.
In the Setup 3-D Surface Parameters dialog box that opens, select the option:
To specify density points, select the option:
To specify surface representation, select the option:
To specify the type of dot symbol, select the option:
2. Run the electrostatic potential.
From the Calculate Electrostatic Potential palette, select Run and Wait. In the File Librarian dialog box that opens, enter the text:
Click the Save button. The electrostatics calculation starts and the cursor changes to a watch. You can view the calculation status in the textport. When the calculation is completed, the cursor returns to an arrow.
From the Calculate Electrostatic Potential palette, select Display Results. The Display Potentials palette and a File Librarian dialog box are displayed.
Select the points.epo file from the File Librarian and then click the Open button.
From the Display Potentials palette, select Show 1-Molecule Potential. In the dialog box that opens for entering an object label, enter the name:
and click the OK button. A dot surface appears in several colors over the cyclohexanol structure. A color-coded potential values scale (in kcal/mol) is displayed in the lower-right corner of the viewing area.
In the Display Potentials palette, select Set Contour Levels. A dialog box offers a set of eight Potential Value data entry fields. The minimum and maximum potential values (in units of kcal/mol) for the simulation are reported at the top of the dialog box. These values can be used to create more realistic contour values.
Enter the value for the Minimum Value of Potential in the Potential Value 1 data entry field. Enter the value for the Maximum Value of Potential in the Potential Value 8 data entry field. Click the Auto Scale button:
Click the OK button to display the contours using the new values.
From the Display Potentials palette select Finish. The Display Potentials palette is replaced by the Calculate Electrostatic Potential palette.
Select the Displayed column for potential2 in the Object Management Table. The dot surface is removed from the viewing area. Objects potential1 and potential2 are both hidden but still attached to the cyclohexanol structure.
A 2D isoenergetic contour plot can be calculated and displayed using a method similar to that for generating grid maps and dot surfaces.
Complete the following exercise to calculate and display this type of object.
1. Set up a 2D contour plot for cyclohexanol.
From the Calculate Electrostatic Potential palette, select Calculate 2-D slice, Calculate from Partial Charges, and Define Plane.
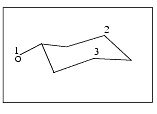
The message line prompts you to pick three atoms in the molecule to define the plane of interest:
From the Select palette, select the cyclohexanol atoms 1, 2, and 3, as indicated in the figure below. When the third atom is selected, the structure is reoriented so the selected plane is parallel to the screen. The Select palette is removed from the screen.
From the Calculate Electrostatic Potential palette, select Run and Wait. A box requests that you specify the atom upon which the zero plane will be defined.
Click the Interactive button and the message line reads:
Pick one atom - pick any palette tool to quit.
Pick atom instead of typing.
Pick one atom to define the center of the plane. When you make your pick, a File Librarian dialog box requests the name for a new output file.
Click the OK button and the calculation proceeds. The cursor appears as an arrow to a watch until the calculation is complete.
Select Display Results from the Calculate Electrostatic Potential palette and a File Librarian dialog box opens.
Select the file 2dpartial.epo and click the Open button. A dialog box asks you to save or reject the current model. Select Save.
From the Calculate Electrostatic Potential palette, select Show 1-Molecule Potential. In the File Librarian dialog box that opens, enter the name potential3 and click the OK button. A 2D contour plot appears in several colors. A color-coded potential values scale (in units of kcal/ mol) is displayed in the lower-right corner of the viewing area.
In the Display Potentials palette, select Set Contour Levels. A dialog box offers a set of eight Potential Value data entry fields. The minimum and maximum potential values (in units of kcal/mol) for the simulation are reported at the top of the dialog box. These values can be used to create more realistic contour values.
In the Potential Value 1 data entry field, enter the value for the Minimum Value of Potential. In the Potential Value 8 data entry field, enter the value for the Maximum Value of Potential. Click the Auto Scale button.
Click the OK button to display the contours using the new values.
5. Hide the 2D contour plot object.
From the Display Potentials palette select Finish. The Display Potentials palette is replaced by the Calculate Electrostatic Potential palette.
Select the Display column for 2dpartial in the Object Management Table. The text changes from no to yes and the 2D contour plot is removed from the viewing area. No objects are displayed with cyclohexanol.
Electrostatic field calculations are performed using the fieldcalc program developed by F. Blaney. The electrostatic field is calculated using essentially the same procedure as employed for the electrostatic potential. The calculated field is shown as colored arrows.
Complete the following exercise to become familiar with the procedure for calculating and displaying an electrostatic field.
1. Set up an electrostatic field calculation.
Display the Calculate menu and select Electrostatic. From the pull- right menu that opens, select Field.
From the Calculate Electrostatic Field palette, select Calculate 2-D Slice, Calculate from Partial Charges, and Define Plane.
The message line prompts you to select the three atoms that define the plane on which the electrostatic field is displayed:
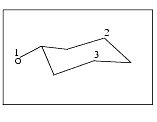
From the Select palette that is displayed, select the cyclohexanol atoms 1, 2, and 3, as indicated in the figure above. When the third atom is selected, the structure is reoriented so the selected plane is parallel to the screen. The Select palette is removed from the screen.
From the Calculate Electrostatic Field palette, select Run and Wait. A dialog box requests that you specify the atom upon which the zero plane will be defined.
Click the Interactive button and the message line reads:
Pick one atom - pick any palette tool to quit.
Pick atom instead of typing.
Pick one atom to define the center of the plane, and a File Librarian dialog box requests the name for a new output file.
When the calculation is complete, select Display Results from the Calculate Electrostatic Field palette. In the File Librarian dialog box that opens, select the file 2dfield.efo and click the Open button. A dialog box asks you to save or reject the current model.
Select Save and the Display Field palette allows you to modify vector arrow size and exclude small vectors to simplify the plot.
Select Display Field. In the File Librarian dialog box that opens, enter the name field1 and click the OK button. A vector field of arrows of different size, color, and direction are displayed. Larger arrows indicate greater field magnitudes, with a blue arrow indicating a negative field and a red arrow indicating a positive field.
From the Display Field palette, select Finish. From the Calculate Electrostatic Field palette, select Finish. The Modeling palette is displayed.
Select the title cell for the Delete column in the Object Management Table. The vector field is removed from the viewing area and all objects are deleted from the Object Management Table. The table closes.
Although hydrogen bonds are generated and displayed like the graphical objects discussed in other sections of this chapter, hydrogen bonds are handled quite differently.
The hydrogen bond function searches a displayed structure for possible hydrogen bonds and monitors the interaction of a molecular structure with a substrate. In the QUANTA Calculate menu, Hydrogen Bond calculates and displays hydrogen bonds any time the structure is changed. The presence or absence of a hydrogen bond is evaluated by whether the prospective atoms meet defined distance and angle criteria.
In the Modeling palette, Hydrogen Bond performs a one-time calculation of hydrogen bonds for the displayed structure and displays the results. Hydrogen bonds are indicated by dashed white lines between the involved atoms.
The hydrogen bond function relies on information in the atom parameter file that indicates whether a particular type of atom is a potential hydrogen bond donor or acceptor. The parameter file contains a hydrogen bond column for each atom type, consisting of one of four letters that indicate the hydrogen-bonding capability of a particular atom type. The letters are:
A polar hydrogen atom is flagged as a donor. When hydrogen atoms are not included in atom data files, atoms attached to polar hydrogen atoms are flagged as donors.
In checking hydrogen bond geometry, the hydrogen bond function considers the donor and acceptor atoms as well as the atoms bonded to the donor and acceptor. Criteria used in checking geometry are:
The limit values can be listed and changed using Bond Options in the Edit menu. Parameters are designated as: d1don, d2don, d1hyd, d2hyd, ang1d, angla, and anglh.
The default values for these parameters are shown in Table 33.
The distance criteria for d2don and d2hyd are greater than for d1don and d1hyd. These criteria are used as the limit for near- hydrogen bonds to be listed to a file. Near-hydrogen bonds are potential hydrogen bonds that do not satisfy the strict criteria to be hydrogen bonds, but have appropriate chemical groups sufficiently close to the distance and angle limits to be considered possible hydrogen bonds. The d2don parameter is used in the same situation as d1don.
The default parameters used in QUANTA are based on the results of the analysis of hydrogen bonding in proteins. See citation 5 in the Reference section at the end of this chapter.
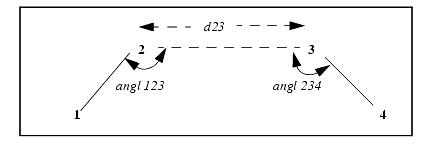
All hydrogen bonds, including near-hydrogen bonds and changes in defaults, can be displayed on screen and listed to a file. In the file, QUANTA lists the atoms used in calculating the hydrogen bond geometry, the angles (see angl 123 and angl 234 in the figure below), and distances (see d23 below). If all atom are not defined, some angles and distances are not calculated.
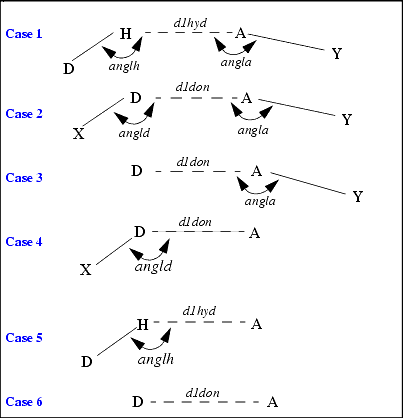
Various situations are possible for hydrogen bonding. For example, the atoms attached to a donor or acceptor may not be defined. The next figure illustrates several situations where hydrogen bonding may occur.
In this figure, atoms are labeled:
The interatomic distances and angles that are calculated in the geometry checking are indicated and labeled with the name of the limiting value.
In Cases 1 and 2, four atoms define the geometry. In Case 1, the hydrogen atom is defined. In Case 2, it is not. In both these cases, a distance criterion and two angle criteria are applied.
In Cases 3 through 5, three atoms define the hydrogen bond geometry. Therefore, only one bond angle criterion can be applied.
In Case 6, only two atoms are defined. Therefore, only the distance criterion can be applied.
Complete the following exercise to become familiar with procedures to calculate, display, and save hydrogen bonds. Bond angle and distance criteria are changed, which changes the number of hydrogen bonds calculated.
Use mypeptide.msf for this exercise. The structure was constructed in the section, Creating a Peptide, in Chapter 4. If you did not complete this section, do so before proceeding.
Display the File menu and select Open. In the Open MSF File Librarian dialog box that opens, select mypeptide.msf from the scrolling list.
Click the Open button to display the structure in the viewing area.
2. Start the Hydrogen Bonds application.
Display the Calculate menu and select Hydrogen Bonds. The Hydrogen Bonds palette is displayed. See Table 33 for a list of the selections in this palette and a brief description of each.
3. Set the hydrogen bond parameters.
From the Hydrogen Bonds palette, select Set Parameters. In the Set up hbond parameters dialog box that opens, enter the values:
Max distance between hydrogen atom and acceptor: 2.5
As above but for "near" hydrogen bonds: 3.0
Max distance between atom bearing hydrogen and acceptor: 3.3
As above but for "near" hydrogen bonds: 4.0
Minimum angle at the acceptor: 90.0
Minimum angle at the hydrogen: 90.0
Minimum angle at the atom bearing the hydrogen: 90.0
4. Calculate and display hydrogen bonds.
From the Hydrogen Bonds palette, select Calculate Hbonds. Dashed white lines connect atoms that are calculated to meet the default distance and geometry criteria set up in the hbond parameters dialog box. Show Hbonds is automatically selected.
From the Hydrogen Bonds palette, select Show Hbonds to turn it off. The dashed lines disappear and the selection is no longer highlighted.
6. Write the list of hydrogen bonds to a file.
From the Hydrogen Bonds palette, select List to File. A File Librarian dialog box prompts you for a filename. Enter the name: mypeptide.
Click the Save button and a dialog box asks for a title for the hbond file. Click the OK button to indicate there is no title for this file. The hydrogen bond information is stored in mypeptide.hbo.
The list of hydrogen bonds is written out to an ASCII file:
Each line of this file reports a different hydrogen bond. The atoms defining the donor are listed first, followed by the acceptor atom(s).
An & symbol in column 3 signifies that the hydrogen bond is in the "near" list.
SEG - the segment name of the donor atom.
RES - the residue name of the donor atom.
ATOM1 - the name of the connecting atom in the donor group (Atom 1).
ATOM2 - the name of the donor atom in the donor group (Atom 2.)
ATOM3 - the name of the acceptor atom in the acceptor group (Atom 3).
ATOM4 - the name of the connecting atom in the acceptor group (Atom 4). (Atom 4 is not present for hydrogen bonds to waters). (Atom 1 is not present for hydrogen bonds from waters).
RES - the residue name of the acceptor atom.
SEG - the segment name of the acceptor atom,
A123 - the angle made between Atom1-Atom2-Atom3,
D23 - the distance between Atom2-Atom3,
A234 - the angle made between Atom2-Atom3-Atom4,
ACON - the angle made between Carbon, Nitrogen and Oxygen when they are in the Hbond (this is an additional measure emulating no- hydrogens on the protein).
DON - the distance between the oxygen and nitrogen (equivalent to ACON).
AHNO - the angle between the hydrogen, nitrogen and oxygen in a hydrogen bond.
The remaining symbols indicate whether the donor/acceptor are symmetry generated (S) or not ( ).
7. Display the hydrogen bond list on screen.
From the Hydrogen Bond palette, select List to Screen. The hydrogen bonds that meet the default criteria are reported in the textport.
8. Change the hydrogen bond parameters and recalculate.
From the Hydrogen Bonds palette, select Set Parameters. In the Set up hbond parameters dialog box, enter the values:
Max Distance between hydrogen atom and acceptor: 2.1
Minimum angle at the acceptor (Min 0 Max 180): 105
Leave all other parameters at their default values.
From the Hydrogen Bonds palette, select Calculate Hbond and the hydrogen bonds are recalculated.
9. Display the new hydrogen bond list on the screen.
From the Hydrogen Bonds palette, select List to Screen. Fewer hydrogen bonds are calculated as a result of tightening the distance and geometry criteria.
From the Hydrogen Bonds palette, select Finish and a dialog box asks how the hydrogen bonds should be used.
No further hydrogen bond calculation
Click the OK button. The Hydrogen Bonds palette is removed from the screen, and the Modeling palette is redisplayed. Hydrogen Bonds is automatically selected in the Modeling palette, and the hydrogen bonds continue to be displayed.
From the Modeling palette, select Hydrogen Bond. The selection is turned off and the hydrogen bonds are deleted.
When you have established parameters, they can be applied to any structure by selecting Hydrogen Bonds in the Modeling palette.
Complete the following exercise to apply the hydrogen bond parameters to a different structure. The MSF newpeptide.msf was created by modifying the structure in the Selecting Active Atoms section of Chapter 2 in QUANTA Basic Operations. If you did not complete this exercise, do so before proceeding.
1. Open the new structure, newpeptide.msf.
Display the File menu and select Open. In the File Librarian dialog box that opens, select newpeptide.msf from the scrolling list. Select Replace and then select Open to display the structure in the viewing area.
2. Calculate the hydrogen bonds.
From the Modeling palette, select Hydrogen Bonds and the hydrogen bonds are calculated using the last specified parameters. The hydrogen bonds are displayed. See Table 34 for a list of the options and a brief description of each.
Select Hydrogen Bonds again to turn it off. The hydrogen bonds are deleted and Hydrogen Bonds is no longer highlighted.
Graphical objects are displayed in relation to but separate from the structures on which they are based. Although each object is a representation of data related to a structure, it is not part of the MSF. However, using the Object Management Table, you may attach an object to an MSF so that it can be moved and deleted with the structure. The Object Management Table also provides object display, naming, and deletion options. Each graphical object is uniquely identified so that you can manage it throughout a QUANTA session.
Some objects, such as ribbons and grid contour meshes, are generated within QUANTA. Other objects, such as brick maps, Connolly solvent surfaces, and 3D vectors, are generated from results of calculations outside of QUANTA.
Although hydrogen bonds are optionally generated and displayed the same as the graphical objects discussed in this chapter, they are handled quite differently. The hydrogen bond function searches a displayed structure for possible hydrogen bonds and monitors the interaction of a molecular structure with a substrate. In the QUANTA Calculate menu, Hydrogen Bond calculates and displays hydrogen bonds any time the structure is changed.
1. M. Carson and C. E. Bugg, J. Molec. Graphics 4, 121 (1986).
2. P. Evans, Daresbury Laboratory Information Quarterly for Protein Crystallography, 19 (1987).
3. M. L. Connolly, Science 221 709 (1983).
4. P. K. Weiner et al., Proc. Nat. Acad. Sci US 79, 3754 (1982).
5. E. N. Baker and R. E. Hubbard, "Hydrogen bonding in globular proteins" Prog. Biophys. Molec. Biol. 44, 97-197 (1984).
6. C. Giessner-Prettre and A. Pullman, Theoret. Chim. Acta, 25 83-88 (1972).