
This chapter covers potential energy and energy minimization calculations as they are executed in QUANTA using the CHARMm program. Read this chapter to understand basic information about CHARMm, the QUANTA-CHARMm interface, and how potential energy and energy minimization calculations are set up and completed.
For more detailed information about CHARMm, consult CHARMm Principles and the CHARMm Dictionary. For more information on forcefields and their uses, please see Forcefield-Based Simulations.
The exercises in this chapter use structures that were created in an earlier chapter. If you did not create and store the MSFs orange.msf and sodium.msf files, go through the exercises in Chapter 1 and do so. If you need to restart QUANTA, refer to the restart procedure in the Preface.
QUANTA uses the CHARMm program to execute many of the calculations needed for modeling. CHARMm is a general and flexible software application developed and maintained at Harvard University to model the structure and behavior of molecular systems. See the References at the end of this chapter for more information.
A variety of systems, from isolated small molecules to solvated complexes of large biological macromolecules, can be simulated using CHARMm.
CHARMm uses empirical energy functions to describe the forces on atoms in molecules. These functions, plus the parameters for the functions, constitute the CHARMm forcefield. CHARMm uses these functions to rapidly calculate conformational energies, local minima, barriers to rotation, energy surfaces, and time-dependent dynamic behavior.
The CHARMm energy functions include internal coordinate terms and pairwise nonbond interaction terms. The total energy can be expressed by the equation:
| Eq. 1 |
|
The internal energy terms include:
The external (i.e., nonbond) energy terms include:
The extra energy terms include:
The hydrogen-bond function is available but is not included by default in CHARMm calculations.
The standalone CHARMm 31b1 documentation can be found on the web here:
http://www.accelrys.com/doc/life/charmm/31b1/doc/Charmm31.Html
CHARMm uses data from these three sources to calculate energies:
The PSF identifies, by number and type, all atoms contained in the structure. Individual atomic masses and charges are included in this file. Additionally, the PSF lists all intramolecular parameters that contribute to the CHARMm energy function. These parameters include bonds, bond angles, torsion angles, and out-of-plane angles. A PSF is constructed from the data contained in an RTF and the CHARMm parameter file. For more information on PSFs and RTFs, see Chapter 1 of QUANTA Basic Operations.
The CHARMm parameter file defines force constants and reference geometries for each type of atom and interaction contained in structures. Cartesian coordinates are provided to CHARMm or are calculated by CHARMm from the standard geometry data contained in an RTF and the reference values stored in the CHARMm parameter file.
QUANTA provides interactive access to CHARMm through a menu-driven graphical interface. QUANTA allows complex modeling problems to be solved by users with relatively little experience with CHARMm. In addition, QUANTA enables experienced CHARMm users to tap all of CHARMm's inherent flexibility.
Along with sending commands to CHARMm, QUANTA also sends the appropriate molecular definition and Cartesian coordinates required for the requested calculation. The molecular definition is contained in an RTF selected from the $CHM_DATA directory, created by ChemNote or the Molecular Editor, or located in a PSF constructed by QUANTA. The Cartesian coordinates are contained in an MSF.
QUANTA provides an easy mechanism to set up, run, and monitor a variety of CHARMm calculations on structures. Some of these calculations, such as energy, minimization, and dynamics calculations, are explicitly called. These functions are selected from the Modeling palette that is displayed when QUANTA is in the Molecular Modeling mode. Other calculations are started automatically by QUANTA applications such as Sequence Builder.
The CHARMm menu, opened from the menu bar, controls both CHARMm access and calculation setup. Table 17 lists the menu items and provides a brief description of each.
Results from CHARMm calculations are periodically sent back to QUANTA. Some results, such as energies, are displayed in the viewing area and in the textport. If new coordinates are calculated for a structure, these coordinates are incorporated into the MSF and the structure is redisplayed in the viewing area.
You have several options for saving MSF changes that result from CHARMm calculations or other structural modifications. These options are accessed through the dialog box that is displayed when Save Changes is selected in the Modeling palette. The Modeling palette is displayed whenever QUANTA is in Molecular Modeling mode. The options for saving changes are listed and described in Table 18.
A CHARMm empirical energy calculation produces a potential energy value. A single-point energy calculation is useful in comparing conformations, computing thermodynamic properties, calculating interaction energies and forces, and evaluating structures during conformational searching. In addition to energy, CHARMm also calculates the forces on each atom in a molecular system.
When you request an energy calculation, CHARMm is automatically started from QUANTA. It remains running throughout the QUANTA session, accepting requests from QUANTA until it is explicitly stopped or the QUANTA session is ended.
Complete the following exercise to become familiar with the potential energy calculation process.
1. Visually set up orange.msf and sodium.msf for calculations.
In the File menu, select Open to display the File Librarian dialog box.
Select sodium.msf and orange.msf from the scrolling list of MSFs then select the Replace and Open buttons. The structures are displayed in the viewing area.
Select Move Atom from the Modeling palette. Pick the sodium ion and move it near the sulfide group of the organic sulfide molecule using the Translate dials on the Dial Emulator palette.
2. Save the coordinates for the new location of the sodium ion and display distance monitors.
Select Save Changes from the Modeling palette. In a dialog box that opens, select the option:
Create a new generation of sodium.msf
Select Distance from the Geometry palette. Show Distance Monitors is checked and highlighted in the palette by default.
Select the sodium ion and then select one of the three oxygen atoms on the organic sulfide molecule. Repeat this procedure two more times. You see a visual representation of the close ionic bonds that hold sodium and the rest of the molecule together.
Select Distance again to inactivate the selection.
3. Calculate the CHARMm potential energy.
Select CHARMm Energy from the Modeling palette. The tool is checked and highlighted.
CHARMm automatically starts and status information indicating the progress of the CHARMm calculation is displayed on the message line.
Additional status information, similar to the information listed below, is displayed in the textport.
4. Complete the CHARMm calculation.
When the calculation is complete, the potential energy is displayed in the upper-right corner of the viewing area. CHARMm Energy is no longer checked and highlighted on the Modeling palette.
A breakdown of the energy contributions similar to the text below is displayed in the textport. Your results will depend on the calculation parameters you chose and the position of the sodium ion.
The total CHARMm energy is: -32.8911
CHARMm minimization locates the molecular conformation with the lowest potential energy. Minimization is often used to prepare a structure for molecular dynamics.
CHARMm offers a choice of minimization techniques. With these choices, global minimum-energy conformations for small molecules or local minimum-energy conformations for larger molecular systems can be determined. You choose the best minimization technique for your structure. Table 19 lists the choices and describes each.
When you run a minimization, define the minimization method and parameters first by choosing Minimization Options from the CHARMm menu in the menu bar. Table 20 lists the parameters you must define in the dialog box that is displayed when you select Minimization Options.
Run a minimization by selecting CHARMm Minimization from the Modeling palette. If CHARMm is not already running, it is automatically initialized when you select CHARMm Minimization.
The next exercise goes through the process of setting up and running a minimization calculation for the structures in orange.msf and sodium.msf.
1. With the structures displayed in the viewing area, select the minimization technique and parameters.
Display the CHARMm menu and select Minimization Options. A dialog box allows you to select a minimization method and specify associated parameters controlling the calculation.
Number of Minimization Steps: 50
Coordinate Update Frequency: 5
Energy Gradient Tolerance: 0.00001
Energy Value Tolerance: 0.000
Initial Step Size: 0.020
Step Value Tolerance: 0.000
Select the OK button. The minimization technique and associated parameters are defined, and the dialog box is cleared from the screen.
From the Modeling palette, select CHARMm Minimization. CHARMm automatically starts the minimization calculation, using the specified minimization technique and parameters.
Status information is printed in the message line indicating the progress of the CHARMm calculation. Additional status information is displayed in the Textport. Undo Changes, Save Changes, and Reject Changes are activated in the Modeling palette.
As the calculation proceeds, the coordinates representing a new conformation of the displayed structure are periodically sent back to QUANTA. The points at which these coordinates are sent to QUANTA is controlled by the Coordinate Update Frequency option in the Minimization Setup dialog box.
The structure is redisplayed with the new coordinates. In addition, the energy value calculated from these new coordinates is displayed in the upper right corner of the viewing area. The textport displays a minimization log similar to.
When the calculation is finished, the final conformation of the structure is displayed in the viewing area. The final energy is displayed in the viewing area and in the textport.
If the requested Energy Gradient Tolerance or Energy Value Tolerance is not met before reaching the maximum number of minimization steps defined in the Minimization Setup dialog box, minimization calculations can be repeated. A repeat minimization calculation can use the same minimization method and parameters or a new method and new parameters.
Complete the following exercise to continue the minimization process for the structures in orange.msf and sodium.msf.
1. Select a new minimization technique.
Display the CHARMm menu and select Minimization Options. A dialog box allows you to select a minimization method and to specify associated parameters controlling the calculation.
Number of Minimization Steps: 50
Coordinate Update Frequency: 5
Energy Gradient Tolerance: 0.0000
Energy Value Tolerance: 0.0001
Initial Step Size: 0.020
Step Value Tolerance: 0.000
Select the OK button. The minimization technique and associated parameters are defined, and the dialog box is cleared from the screen.
2. Start the second calculation.
From the Modeling palette, select CHARMm Minimization. CHARMm automatically starts the minimization calculation, using the specified minimization technique and parameters.
Status information is printed in the message line and textport.
As the calculation proceeds, the coordinates representing a new molecular conformation of the displayed structure are periodically sent back to QUANTA.
When the calculation is complete, the structure is redisplayed with new coordinates. In addition, the energy value calculated from these new coordinates is displayed in the upper right corner of the viewing area and in the textport.
When a minimization calculation has produced an appropriate structure, the new coordinates of the minimized structure can be saved in an MSF by selecting Save Changes in the Modeling palette.
If you do not want to save minimized coordinates, select Reject Changes in the Modeling palette. When Reject Changes is selected, the minimized structure is replaced by the structure that was last saved to disk. All coordinate changes, whether made by the minimization calculation or by any other calculation, such as Move Fragment, are lost.
The following exercise goes through the process of saving the structures in orange.msf and sodium.msf.
From the Modeling palette, select Save Changes.
A dialog box allows you to reject or save current changes for the file sodium.msf.
Save as a new generation of sodium.msf
Click Save to save the minimization results.
In the dialog box that opens for the orange structure, select the option:
Save as a new generation of orange.msf
Click Save to save the minimization results.
2. Rename and save the structures as a single MSF.
Display the File menu and select Save As. In the Write Options dialog box, select the following write options:
Write out current connectivity
Write out current bondtypes
Use the new MSF now
Select the OK button. In a File Librarian dialog box, enter the name orangeII and click Save. The new file orangeII.msf is displayed in the Molecule Management Table and the textport reads:
35 atoms selected for molecular structure orangeII.msf 35 atoms will be displayed
The Merck Molecular Force Field (MMFF), developed by Halgren at the Merck Research laboratories, is designed for use with a large variety of chemical systems. Its pivotal application is the study of receptor-ligand interactions involving proteins or nucleic acids as receptors and a wide range of chemical structures as ligands. The forcefield can describe ligand and receptor in isolation as well as bound.
MMFF is a computationally derived energy expression that can accurately be applied to condensed-phase and aqueous processes. It uses a unique functional form for describing van der Waals interactions and employs novel combination rules that systematically correlate van der Waals parameters with those that describe experimentally characterized interactions involving rare-gas atoms. The MMFF energy expression is:
| Eq. 2 |
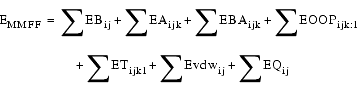
|
EBAijk stretch-bend interactions
EOOPijk;l out-of-plane bending at tri-coordinate centers
Evdwij van der Waals interactions
EQij electrostatic interactions
Constituent terms of the energy expression are calculated in kcal/mol. They are described in detail elsewhere.2,3
To allow straightforward application to condensed-phase simulations employing explicit solvent molecules, MMFF uses a dielectric constant in its electrostatic interaction terms.
1. Select CHARMm mode from the CHARMm menu.
2. Select MMFF from the CHARMm mode pull-right menu. A textport message reads:
Subsequent CHARMm calculations will use MMFF mode.
This chapter describes the basis and procedures for running potential energy and energy minimization calculations. These calculations are executed in QUANTA using the CHARMm program created and maintained by Harvard University.
QUANTA provides interactive access to CHARMm through a menu-driven graphical interface. When you request an energy calculation in QUANTA, CHARMm is automatically started and remains running until it is explicitly stopped or the QUANTA session is ended.
Energy functions plus parameters for the functions constitute the CHARMm forcefield. Much of QUANTA's modeling analysis is based on the CHARMm forcefield.
CHARMm uses data from three sources to calculate energies: a principle structure file (PSF), CHARMm parameters, and Cartesian coordinates for all atoms. A CHARMm empirical energy calculation produces a single potential energy value.
Sequential potential energies and their rate of change are the basis of energy minimization. CHARMm minimization locates the molecular conformation with the lowest potential energy. You may select one or any combination of five methods used in CHARMm for a minimization calculation. The results of a calculation may be accepted and saved or rejected
1. B. R. Brooks, R. E. Bruccoleri, B. D. Olafason, D. J. States, S. Swaminathan, M. Karplus "CHARMm: A program for macromolecular energy, minimization, and dynamics calculations", J. Comp. Chem. 4 187-217.
2. Halgren, T. A., J. Amer. Chem. Soc. 114 7827-7843 (1992).
3. Halgren, T. A., The Merck Molecular Force Field, privately published paper available from Accelrys.
4. Forcefield-Based Simulations, Accelrys, San Diego.