This chapter describes methods available to compare two or more molecular structures by overlaying the structures in the viewing area. Read this chapter if you plan to evaluate molecules for structural similarity.
Structures needed for the exercises in this chapter are built using the 2D Sketcher (ChemNote) application and are minimized using CHARMm. If you are not familiar with ChemNote, read Chapter 2 of QUANTA Generating and Displaying Molecules. Read Chapter 3 of the same book for information on energy minimization procedures.
The Molecular Similarity application provides a visual comparison of two or more molecular structures by overlaying these structures in the viewing area. Comparisons can be made between:
The only requirement is that each structure must be contained in a unique MSF. The application does not permit use of different generations of the same MSF.
The procedure used in Molecular Similarity to compare structures is divided into four basic steps:
1. Load the structures to be compared.
2. Define atom equivalences in these structures, that is, establish atom matches.
3. Perform a fitting operation.
Each structure is identified by the name of its corresponding MSF. One structure is identified as the target or fixed structure. All other structures are working or moving structures. Although only one structure can be designated as the target at a given time, the target definition can be changed from within the application using Define Target Molecule in the Molecule Similarity palette.
The Molecular Similarity application makes no assumptions about the equivalency of atoms on different structures. Equivalency is defined through your input. When atom equivalencies are defined, they can be used for any fitting operation.
Fitting operations employ either a rigid or flexible method. If a rigid method is used, working structures are translated and rotated to obtain an optimum fit with the target structure. If a flexible fitting method is used, conformations of the working structures are changed to obtain an optimum fit.
Fitting operations use a least squares fitting algorithm. The least squares algorithm computes the optimum translation and rotation to be applied to the moving structure such that rmsd of the fit over the specified pairs of equivalent atoms is an absolute minimum. Although the algorithm deals only with a pair of structures at a time, fitting tools available in the application can handle a collection of molecules.
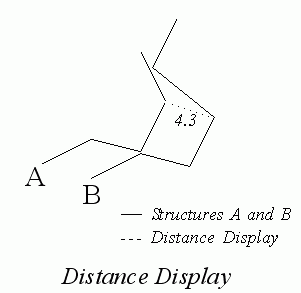
Several methods are provided for analyzing the results of a fitting operation. The target and working structures can be alternately displayed to examine the differences between the overlaid structures. Distance displays that include dashed lines and a numeric value between equivalent atoms on two structures provide indications of the quality of the fit.
Fitted structures can also be analyzed by computing the volume of overlap between the structures. If similarity calculations do not generate an optimal fit between structures, atom equivalences can be modified and fitting operations can be repeated.
In this section, use the 2D Sketcher (ChemNote), accessed through the Applications menu, to create three structures: quinupramine, imipramine, and amitriptyline. Use these structures as test molecules in the exercises that introduce the Molecular Similarity application.
The minimum energy conformation of these structures is determined and saved before fitting operations proceed. The structures imipramine and amitriptyline are minimized using simple CHARMm minimization procedures. The structure quinupramine is a more rigid structure. Run Conformational Search using the Grid Search method to ensure that the lowest energy conformation of quinupramine is found. If you are not familiar with Conformational Search, see Chapters 3-5. When the Grid Search is completed, run Analysis to select and save to the MSF the lowest energy conformation. If you are not familiar with the Analysis applications see Chapter 6.
Complete the following exercise to build and minimize the test molecules.
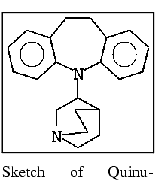
1. Build and save the quinupramine structure.
Display the Applications menu and select Builders. From the pull- right menu that opens, select 2D Sketcher. The ChemNote window is displayed over the QUANTA window.
Sketch the structure quinupramine illustrated below.
Display the ChemNote File menu. Select the Save function. A File Librarian dialog box is displayed.
Enter the name quinpr in the File Librarian name field. Select the Save button. A dialog box is displayed, offering several methods for the adjustment of partial charges.
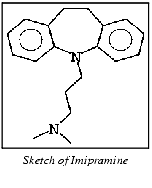
Select the OK button. The dialog box is cleared from the screen. The partial charges are changed, and the new charges are saved in the quinpr.mol file. The sketch remains in the ChemNote window, providing the opportunity to use it as a base for creating a .mol file for imipramine.
Sketch the structure imipramine illustrated below.
Display the ChemNote File menu. Select the Save function. A File Librarian is displayed.
Enter the name imip in the File Librarian name field. Select the Save button. A dialog box is displayed, offering a list of methods for the adjustment of partial charges.
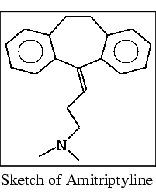
Select the OK button. The dialog box is cleared from the screen. The partial charges are changed, and the new charges are saved in the imip.mol file. The sketch remains in the ChemNote window, providing the opportunity to use it as a base for creating a .mol file for amitriptyline.
3. Build and save amitriptyline.
Sketch the structure amitriptyline illustrated below.
Display the ChemNote File menu. Select the Save function. The File Librarian dialog box is displayed.
Enter the name amit in the File Librarian name field. Select the Save button. A dialog box is displayed, offering several methods for the adjustment of partial charges.
Select the OK button. The dialog box is cleared from the screen. The partial charges are changed, and the new charges are saved in the amit.mol file. The sketch remains in the ChemNote window.
Display the ChemNote File menu. Select the Return to Molecular Modeling function. The ChemNote window is removed from the screen, revealing the Modeling window.
The file amit.mol is automatically converted to amit.msf in the Molecular Modeling mode. When amit.msf creation is completed, a dialog box is displayed offering options to save the structure.
Select the OK button. The structure amitriptyline is displayed.
4. Energy minimize amitriptyline.
Display the CHARMm menu and select Minimization Options. A dialog box is displayed in which you can select a minimization technique and specify associated parameters.
Select the OK button. The dialog box is cleared from the screen. The new minimization parameters are established. The steepest descents method is chosen primarily to improve conformation.
From the Modeling palette, select CHARMm Minimization. The CHARMm calculation is started. The cursor changes from an arrow to a watch. Minimization is completed when the watch is restored to an arrow. The minimized energy results are displayed in the upper- right corner of the viewing area and in the textport.
5. Change parameters and continue minimizing amitriptyline.
Display the CHARMm menu and select the Minimization Options function. A dialog box is displayed in which you can select a minimization technique and specify associated parameters.
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. New minimization parameters are established.
Select CHARMm Minimization. The minimization process proceeds using the new parameters. It stops when the rms force reaches zero.
6. Save minimization results in a file.
From the Modeling palette, select Save Changes. A dialog box is displayed, permitting the new coordinates of amit.msf to be saved.
Select the OK button. The new coordinates replace the original coordinates in amit.msf.
7. Convert imip.mol to an MSF.
Display the File menu and select Import. A File Librarian dialog box is displayed listing the available external file formats and the files available for the selected format
From the bottom scrolling list, select the data format:
The upper scrolling list displays the .mol files that are present in the current directory.
Select the file imip.mol from the scrolling list.
Select the Import button. When the creation of imip.msf is completed, a dialog box is displayed offering the option to use the structure just created.
Select the OK button. The structure imipramine replaces the display of amitriptyline in the viewing area.
Display the CHARMm menu and select Minimization Options. The dialog box containing minimization techniques and parameters choices is displayed.
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. New minimization parameters are established.
From the Modeling palette, select CHARMm Minimization. The CHARMm calculation is started.
When minimization is complete, display the CHARMm menu again. Select Minimization Options. The dialog box containing minimization techniques and parameters choices is displayed.
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. New minimization parameters are established.
From the Modeling palette, select CHARMm Minimization. The minimization process proceeds, using new parameters. It stops when the rms force reaches zero.
9. Save minimization results in a file.
From the Modeling palette, select Save Changes. A dialog box is displayed offering options to save the new coordinates of imip.msf.
Select the OK button. New coordinates replace the original coordinates in the MSF.
10. Convert quinpr.mol to an MSF.
Display the File menu and select Import. A File Librarian dialog box is displayed listing the available external file formats and the files available for the selected format
From the bottom scrolling list, select the data format:
The upper scrolling list displays the .mol files that are present in the current directory.
Select the file quinpr.mol from the scrolling list.
Select the Import button. When the creation of quinpr.msf is completed, a dialog box is displayed offering the option to use the structure just created.
Select the OK button. The structure quinupramine replaces the display of imipramine in the viewing area.
Display the CHARMm menu and select Minimization Options. The dialog box containing the minimization techniques and parameters is displayed.
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. The new minimization parameters are established.
From the Modeling palette, select CHARMm Minimization. The CHARMm calculation is started.
When the minimization is completed, display the CHARMm menu and again select Minimization Options. The dialog box containing the minimization techniques and parameters is displayed.
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. New minimization parameters are established.
From the Modeling palette, select CHARMm Minimization. The minimization process proceeds using new parameters. It stops when the rms force reaches zero.
12. Save minimization results in a file.
From the Modeling palette, select Save Changes. A dialog box is displayed, permitting you to save the new coordinates of quinpr.msf. Select the option:
Select the OK button. New coordinates replace the original coordinates in the MSF.
13. Start Conformational Search.
Display the Applications menu and select Conformational Search. The Conformational Search palette replaces the Modeling palette and supplements the display of the Geometry palette.
14. Define torsions for the search.
From the Conformational Search palette, select Torsions. The Torsions palette supplements the display of the Conformational Search and Geometry palettes.
From the Torsions palette, select Define All Torsions.
From the Torsions palette, select Exit Torsions. The palette is removed from the screen.
15. Select the Grid Scan search method and define specifications.
From the Conformational Search palette, select Setup Search. All of the search method selections become active. Search Setup is checked and highlighted.
From the Conformational Search palette, select Grid Scan. A dialog box is displayed allowing you to edit the values that define rotational range and torsion angle increments within this range.
Select the OK button. A dialog box is displayed, offering options for processing each conformation.
Select the OK button. The dialog box is cleared from the screen. Grid Scan remains checked and highlighted. All other method selections are dimmed.
16. Setup minimization for the search.
Display the CHARMm menu and select Minimization Options. The dialog box containing the minimization techniques and parameters is displayed
Select the minimization option:
Select the OK button. The dialog box is cleared from the screen. New minimization parameters are established.
17. Start the Grid Scan search.
From the Conformational Search palette, select Do Search. The selection is checked and highlighted. A File Librarian dialog box is displayed.
Enter the name quinpr for the file that holds the search results.
Accept default values for tag specifications:
Select the New button. The search procedure is started. New conformations are displayed in the viewing area every few seconds. When the search is completed, the last conformation is displayed in the viewing area and the message line displays:
From the Conformational Search palette, select Analysis.
The Conformational Search application is temporarily exited. The Analysis and Plots palettes replace the Conformational Search palette. The Geometry palette remains in view.
The first conformation from the search is displayed. The conformation is labeled in the upper-left corner: quinpr_msf:1. There is also an informational line indicating the total number of conformations generated in the search and the number that is currently selected. The line reads:
19. Display the conformation with the lowest potential energy.
From the Analysis palette, select Lowest Energy Structure. The conformation is displayed in the viewing area along with its associated energy and label. The actual conformation may vary, depending on the initial energy of the minimized structure.
From the Analysis palette, select File Options/Filters. A dialog box is displayed offering the option to write the coordinates of the displayed conformation to an MSF.
Select the OK button. A File Librarian dialog box is displayed.
Select quinpr.msf from the scrolling list.
Select the OK button. A dialog box is displayed asking if it is OK to overwrite the existing file.
Select the Yes button. The low energy conformation replaces existing coordinates in quinpr.msf.
21. Exit Analysis and Conformational Search.
From the Analysis palette, select Exit Analysis. The Analysis and Plots palettes are removed from the screen and the Conformational Search palette is redisplayed.
From the Conformational Search palette, select Exit Conformational Search. The Conformational Search palette is removed from the screen and the Modeling palette is redisplayed.
22. Reject the changes made during the search.
From the Modeling palette, select Reject Changes. All modeling changes made during the search and minimization are discarded. The lowest energy conformation as a result of the search remains saved in the file quinpr.msf.
The Molecular Similarity palette provides selections to:
Rotation, translation, and display selections in the Molecular Similarity palette are used during atoms picking and after fitting procedures to obtain a clearer display of structures. If additional fitting operations are needed to resolve other similarity questions, superimposed structures can be separated and the target structure assignment can be changed.
Other palette selections are used to manage the files that are generated during fitting and comparison operations. Table 35 lists and briefly describes all palette selections.
The Molecule Management Table is used to change the activity and display of structures. Clicking on the activity column of a molecule in the table toggles the activity of the molecule. Molecule activity can also be changed by clicking on any atom of a molecule in the viewing area.
The activity toggle can be disabled by selecting Options in the Molecule Similarity palette, opening the Other Options dialog box, and deselecting Enable Atom Picks to Control Molecular Activity. If this feature is enabled, a click on an atom is equivalent to clicking on the activity column of the Molecule Management Table. The molecular display momentarily highlights whenever a molecule's activity changes.
The display of a molecule in the viewing area may be toggled from visible to invisible, or the reverse, by clicking on the appropriate cell of the Visible column for the molecule of interest.
Take time to note areas of the structures you are studying that appear to be similar. This will help you identify candidates for atom matching procedures. To make them easy to locate, label candidate atoms in each structure using Label Atoms in the Draw menu. Also use Bond Style in the Draw menu to alter color and vector representations of bonds, so that double and triple bonds are easy to identify.
Complete the following exercise to prepare amitriptyline and quinupramine for atom matching.
Display the File menu and select the Open function. A File Librarian dialog box is displayed.
Select amit.msf from the scrolling list and then select the Replace button. Select the Open button. Amitriptyline replaces quinupramine in the viewing area.
2. Display the multiple bond in amit.msf.
Display the Draw menu and select Bond Style. From the pull-right menu that opens, select Draw Half-bond Vectors. Make a second selection, Multi-vector Bonds. The two aromatic rings and the double bond are now clearly visible in the viewing area.
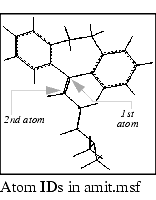
3. Identify atoms at the double bond endpoints.
Pick the atom at each end of the double bond. An ID is displayed for each atom. Note the names of the two atoms, so that you can identify the double bond during fitting procedures.
Display the File menu and select the Open function. A File Librarian dialog box is displayed.
Select quinpr.msf from the scrolling list and then select the Append button. Select the Open button. Quinupramine and amitriptyline both appear in the viewing area. In the Molecule Management Table, the status of both structures is indicated as visible and active.
5. Start Molecular Similarity.
Display the Applications menu and select Molecular Similarity. The Molecular Similarity palette replaces the Modeling palette and supplements the Geometry palette.
6. Space structures in the viewing area so they do not overlap.
Display the View menu and select Stamp. This selection divides the viewing area into several smaller viewing areas. Each of the areas is stamped with only one molecule so that the structures in the viewing area no longer overlap.
In the Molecule Management Table, select the Active cell for amit.msf to make it inactive.
From the Molecular Similarity palette, select Rotate Molecules in Place. The dial emulators change to provide task-specific dials for x, y, and z rotations.
Select the X Rotation dial. Since quinupramine is the only active structure in the Molecule Management Table, it is the only structure to rotate about the x-axis.
Make quinupramine inactive by clicking on the structure's activity cell in the Molecule Management Table. Make amitriptyline active by clicking on its activity cell in the Molecule Management Table.
Select the Y Rotation dial. Amitriptyline rotates about the y axis.
Make quinupramine active again. Select the Z Rotation dial. Since both structures are now active, they both rotate about the z axis.
Select Rotate Molecules in Place to deselect it. The task-specific dials for x, y, and z rotation are replaced with Dial Set 1.
9. Control the display of quinpr.msf.
Click on the Visible cell for quinupramine in the Molecule Management Table. The structure is no longer displayed in the viewing area although it is still marked active in the table. Click again on the same Visible cell. Quinupramine is redisplayed.
10. Exit Molecular Similarity.
From the Molecular Similarity palette, select Exit Molecular Similarity. The program returns to Molecular Modeling mode.
In Molecular Similarity, the first-loaded structure is identified by default as the target structure. The target molecule has an * after its name in the Molecule Management Table. All other structures are working structures.
The target molecule is changed by selecting a row in the Molecule Management Table, then selecting Define Target Molecule on the Molecular Similarity palette.
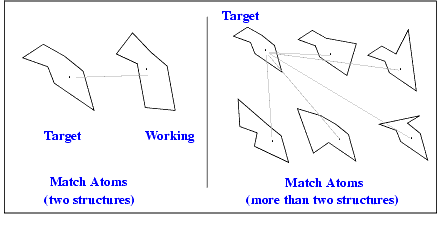
The Match Atoms procedure links equivalent or near equivalent atoms on one or more working molecules to the corresponding atom in the target structure. Atom equivalences are always defined in pairs. When you use multiple working structures, equivalent atoms are individually paired with the target atom, not collectively grouped with each other. The following figure illustrates these matches.
With matches established, relationships between target and working atoms are defined, permitting sophisticated structural comparisons.
An atom equivalence list containing atom pairs is required before any type of fitting operation can be performed. A list is automatically created by picking equivalent atoms in the displayed structures. The tools needed for defining this list are contained in the Match Atoms palette. This palette opens when Match Atoms is selected from the Molecular Similarity palette. Table 36 lists and briefly describes the palette selections.
With two structure involved in a matching procedure, there are two methods available to establish atom matches:
When more than two structures are involved in atom matching, a modification of the second matching method is used.
Complete the following exercise to become familiar with procedures for matching atoms in two structures. In this exercise, the structure amitriptyline is designated as the target structure and the structure quinupramine is designated as the working structure.
1. Start the Molecular Similarity application.
Display the Applications menu and select Molecular
Similarity.
2. Set up Molecular Similarity options.
From the Molecular Similarity palette, select Options. A dialog box is displayed.
Select the OK button. The Set Other Options for Molecular Similarity dialog box is displayed.
3. Rotate structures until their views are parallel.
From the Molecular Similarity palette, select Rotate Molecules in Place. The Dial Emulator changes to provide task-specific dials for x, y, and z rotations for active molecules.
Use a combination of Active cell choices and x, y, and z dial rotations to adjust the position of each structure until the view of one structure is parallel to the view of the other, as in the illustration. The similarity of structure positions assists in locating equivalent atoms during atom matching procedures.
4. Deselect Rotate Molecules in Place.
From the Molecular Similarity palette, select Match Atoms. The Match Atoms palette supplements the display of the Molecular Similarity and Geometry palettes.
From the Match Atoms palette, select Pick Equivalent Atoms.
The textport displays instructions for picking atom matches:
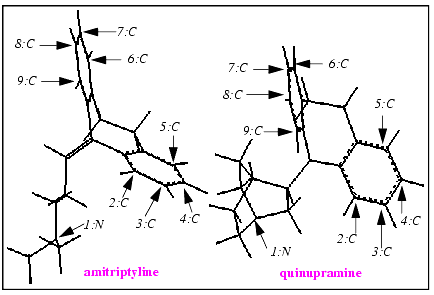
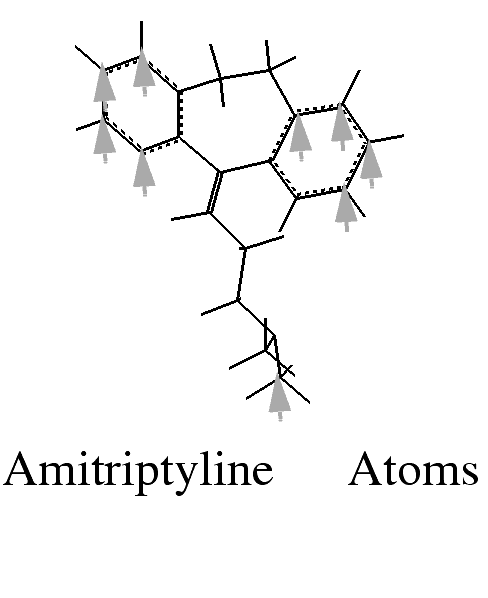
From the amitriptyline molecule, pick the nitrogen atom indicated in the illustration above. The atom ID is displayed. Note that incorrect choices can be reversed by using Undo Last in the Match Atoms palette.
Pick the indicated equivalent nitrogen atom in quinupramine. The atom ID is displayed along with a dashed line to the nitrogen atom in amitriptyline.
Continue picking equivalent atoms until nine pairs are selected. Rotate structures as needed to see the atoms to be picked. Dotted lines are drawn to indicate all atom matches.
From the Match Atoms palette, select End Atom Picking. Pick Equivalent Atoms is turned off. The textport reports the number of atoms matched:
From the Match Atoms palette, select List Atom Matches. A list of matches is displayed in the textport. As in the example below, the first column in this list indicates the number associated with that atom match. The second and third column indicate the atoms composing these matches. The fourth column indicates distance between matched atoms. The rms distance deviation over all atom matches is indicated at the end of the list.
Actual atom names may differ, depending on the sequence in which bonds and atoms were placed when the structures were created in ChemNote. Actual rms deviation will differ if the degree of minimization for the structures was not the same.
From the Match Atoms palette, select Exit Match Atoms. The Match Atoms palette is removed from the screen.
From the Molecular Similarity palette, select Exit Molecular Similarity. The Molecular Similarity palette is replaced by the Modeling palette.
9. Reject the changes made from the Manage Atoms palette.
From the Modeling palette, select Reject Changes. The changes are discarded to prepare for matching atoms in three structures.
When there are more than two structures for atom matching, alternately selecting match atoms between target and working structures is no longer possible. When three or more structures are involved, matching atoms are selected in the following manner:
1. The target structure is displayed in the viewing area by itself in an enlarged size. Matching atoms on the target structure are picked.
2. The first working structure is displayed with the target structure. Corresponding atoms on the working structure are picked in the same order they were chosen on the target structure.
3. The second working structure is displayed. Corresponding atoms on this structure are picked in the same order they were chosen on the target structure.
4. The corresponding atoms on any additional structures are picked, one structure at a time.
Complete the following exercise to become familiar with the procedure for matching atoms on three or more structure. The equivalent atoms established in this exercise are used in the fitting method section of this chapter. You are automatically prompted through the steps in this procedure.
1. Open quinpr.msf, amit.msf, and imip.msf, changing the target.
Display the File menu and select Mono. When the files are opened, the structures are displayed one on top of the other.
Display the Draw menu and select Open. A File Librarian dialog box is displayed.
Select quinpr.msf, amit.msf, and imip.msf from the scrolling list. The structures will be displayed in the order selected. Quinupramine is the target molecule.
Select the Replace button and then the Open button. All three structures are displayed stacked on top of each other. The Molecular Management Table indicates all three structures are active and visible.
2. Start Molecular Similarity.
Display the Applications menu and select Molecular Similarity. The Molecular Similarity palette replaces the Modeling palette and supplements the display of the Geometry palette.
3. Setup Molecular Similarity options.
From the Molecular Similarity palette, select Options. A dialog box is displayed.
Select the OK button. A second dialog box is displayed.
Accept all other defaults selections and the default values:
From the Molecular Similarity palette, select Match Atoms. The Match Atoms palette appears.
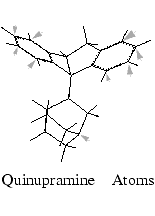
From the Match Atoms palette, select Pick Equivalent Atoms. Only quinupramine is visible in the viewing area.
Use the Dials palette to orient and scale the structure for atom picking.
Click on each atom that is selected to match it with equivalent atoms on amitriptyline and imipramine. Refer to the illustration for designated atoms. As picking proceeds, a colored circle is placed on each picked atom.
When all nine atoms have been picked, select End Atom Picking from the Match Atoms palette.
5. Select matching atoms from amitriptyline.
Amitriptyline reappears in the viewing area beside quinupramine. The first quinupramine atom requiring a matching atom has an enlarged crosshair placed on the circle that marks it.
Pick the corresponding atom from the amitriptyline structure. When the equivalent atom is picked, the large crosshair shifts to the next atom in the target molecule. Select the matching atom in the working molecule. Repeat this procedure until all nine matching atoms on amitriptyline have been picked. When the last matched pair is selected, the quinupramine match atoms are no longer highlighted. Amitriptyline is removed from the viewing area and quinupramine and imipramine are both displayed.
6. Select match atoms from imipramine.
Select the imipramine match atoms in the same manner as for amitriptyline match atoms. When all match atoms are chosen, all three structures are displayed in the viewing area, with equivalent atoms on all structures connected by dashed lines.
Pick Equivalent Atoms is turned off and the textport reports the number of atoms matched:
From the Match Atoms palette, select List Atom Matches. A list of matches is displayed in the textport. The first column in this list indicates the number assigned to each match. The second and third columns indicate the atoms composing the matches The fourth column indicates the rms distance difference for each match, while the overall rms distance deviation is indicated at the end of the list.
1 N34:1 N30:2 9.28
2 C23:1 C20:2 9.54
3 C22:1 C21:2 9.56
4 C21:1 C22:2 9.35
5 C20:1 C23:2 9.20
6 C15:1 C12:2 9.47
7 C14:1 C13:2 9.78
8 C13:1 C14:2 9.94
9 C12:1 C15:2 9.79
Actual atoms names may differ depending on the sequence in which bonds and atoms were placed in ChemNote when the structures were created.
From the Match Atoms palette, select Exit Match Atoms. The equivalent atoms established in this exercise are used in the fitting methods section that follows.
There are two types of fitting procedures available: rigid-body fitting and flexible fitting. Rigid-body fitting translates and rotates working structures to minimize the rms of the fit to the target structure. Flexible fitting modifies the conformations of the working structures to improve the quality of the fit.
Two methods of rigid-body fitting are available when an equivalence list is established: rigid body fit to common frame and rigid body fit to target.
Rigid Body Fit to Common Frame in the Molecular Similarity palette performs an optimal overlay of structures when no one particular structure has a unique role as target molecule. A common frame is created that consists of a set of points corresponding to mean positions of all matched atoms. The structures are then fit to this frame with the frame acting as the target structure. The fitting procedure is repeated each time, re-evaluating the common frame.
Rigid Body Fit to Target in the Molecular Similarity palette fits each active working structure to the target structure, providing a superimposed display of structures in the viewing area. The rms difference of the working structures, with respect to the target structure, is reported in the textport. The overall rms deviation (the sum of the rmsd of each working structure divided by the rmsd of the target structure) is reported.
There are also two methods of flexible fitting: torsional flexible fit and Cartesian flexible fit.
Torsional Flexible Fit in the Molecular Similarity palette manipulates the conformation of each working structure by suitably modifying its rotatable torsion angles. The target structure remains fixed. As the conformation is modified, it is rigid body fitted to the target, and the rms difference of the fit is evaluated. The rms difference is then minimized with respect to the torsion angles of the working structure. The rms minimization procedure is performed for one working structure at a time.
Cartesian Flexible Fit in the Molecular Similarity palette uses a CHARMm energy minimization with target constraints to achieve a fit of the working structures to the target structure. Through this process, the working structure is forced to conform to the conformation of the target structure, that is, the distances between matched pairs of atoms are effectively reduced to zero. After minimization, all target constraints are cleared.
Complete the following exercise to become familiar with fitting procedures.
1. Fit amitriptyline and imipramine to the target structure quinupramine.
From the Molecular Similarity palette, select Rigid Body Fit to Target. The structures amitriptyline and imipramine are superimposed over the structure quinupramine, using picked equivalent atoms as the overlay points.
The textport reports the rms difference of each structure, with respect to the target structure, and the overall rms deviation.
2. Review results of the fitting procedure in the Textport.
Select the Reset View dial emulator. The structures are repositioned in the viewing area.
From the Match Atoms palette, select List Atom Matches. The textport reports the rms distances for each atom pair and the overall rms deviation.
The first column indicates the number of the atom pair. The second column indicates the picked atom in the target structure, and the third column indicates the equivalent atom picked in the first working structure. The fourth column (labeled 2) indicates the distance between the pair of atoms. The fifth column shows the equivalent atom in the next working structure, while the last column (labeled 3) indicates the distance of this atom from the equivalent atom in the target structure. The overall RMS deviation is the same value that was reported in the brief Textport summary at the completion of the fitting procedure.
3. Analyze visual results of the fitting procedure.
Dashed lines between equivalent atoms are displayed by default. Display Atom Matches is checked and highlighted in the Match Atoms palette.
From the Match Atoms palette, select Display Distances. The distance between equivalent atoms is displayed next to these atom locations in the viewing area.
From the Match Atoms palette, select Display Distances. The distance between equivalent atoms is displayed next to these atoms in the viewing area. The Match Atoms palette is cleared from the screen.
4. Define torsions for the torsional flexible fit procedure.
From the Molecular Similarity palette, select Torsions... The Torsions palette is displayed. Check to see that Define All Torsions is displayed. From the Torsions palette, select Define All Torsions. The message line displays:
Torsion definitions are reported in the textport.
From the Torsions palette, select Select Torsions. The Torsion Selection palette is displayed.
From the Torsion Selection palette, select Select a Subset of Torsions. A dialog box containing a list of all defined torsions is displayed. All torsions are selected.
5. Exclude the torsion associated with the double bond in amitriptyline.
Deselect the torsion that includes the carbon atoms you marked in Step 3 of the exercise in Managing Structures on page 197.
Select the OK button. The dialog box is cleared from the screen.
Display the Torsion Selection palette and select Finish. The palette is cleared from the screen.
From the Torsions palette, select Exit Torsions. The palette is cleared from the screen.
6. Perform a torsional flexible fit procedure.
Display the Molecular Similarity palette and select Torsional Flexible Fit. Structures are displayed as the fitting procedure is performed. The rms distances between each structure and the target is reported in the textport, and the overall rms deviation is shown, at the completion of the procedure.
7. Review the results of the fitting procedure in the textport.
Select the Reset View dial emulator. The structures are repositioned in the viewing area.
Display the Match Atoms palette. Select the tool List Atom Matches. The textport reports the rms distance for each atom pair and the overall rms deviation.
Comparing the results of the torsional flexible fitting with the results of the rigid body fitting shows the RMS values are the same or lower between quinupramine and imipramine (structure 2) and between quinupramine and amitriptyline (structure 3).
8. Setup CHARMm minimization conditions.
Display the CHARMm menu and select Minimization Options. A dialog box is displayed, permitting a minimization technique to be selected and associated parameters to be specified.
Select the OK button. The dialog box is cleared from the screen. The new minimization parameters are established.
9. Perform a Cartesian flexible fit procedure.
From the Molecular Similarity palette, select Cartesian Flexible Fit. The cursor changes to a watch during the fitting operation. The message line shows CHARMm status, and the structures are dynamically positioned in the viewing area as the fitting procedure is performed. The procedure is complete when the cursor returns to an arrow.
From the Molecular Similarity palette, select Save Changes.
10. In the Textport, review the results of the fitting procedure.
Select the Reset View dial emulator. The structures are repositioned in the viewing area.
From the Match Atoms palette, select List Atom Matches. The textport reports the rms fit of each atom pair and the overall rms deviation. An example of the data follows:
The results of the Cartesian flexible fit show that the rms values are reduced to zero or close to zero.
In addition to the visual inspection of overlaid structures, it is possible to calculate the van der Waals volume of structures and display this information in a variety of volume maps.
Three types of volume maps can be displayed:
The union volume map displays all van der Waals volumes calculated for every structure. The intersection volume map displays only the area where structures overlap. The volume density map superimposes a grid over the cumulative union volume map and, through the use of color, displays the proportional number of structures occupying a given grid point.
Additional functions, such as making each structure a different color or alternating the display of structures, can help in the analysis of volume maps. Selections used to calculate, display, and compare volume maps are located in the Molecular Similarity palette.
Complete the following exercise to become familiar with volume calculations and display options.
From the Molecular Similarity palette, select Calculate Volumes. A dialog box is displayed to permit adjustment of the grid resolution.
The dialog box is cleared from the screen and the cursor changes to a watch during the calculation. The textport reports the results of the calculation.
As the calculation proceeds, the Object Management Table is displayed to provide standard object information on each map display.
2. Display volume maps separately.
Select the density1 Visible cell. The cell entry changes from Yes to No. The map showing the total volume of all three structures is removed from the viewing area. The union and intersection maps remain displayed.
Select the union1 Visible cell. The cell entry changes from Yes to No. The map showing the van der Waals volumes calculated for every structure is removed from the viewing area. Only the intersection map remains displayed.
3. Determine the structures that have non-overlapping areas.
Display the Draw menu and select Color Atoms.
From the pull-right Color Atoms menu, select Color by Molecule. Each molecule is displayed in a single color.
Rotate the intersection map and locate areas where bonds and atoms extend beyond the map boundaries. To help determine the molecule that has non-overlapping areas, toggle visibility of each atom on and off.
From the Molecule Management Table, select the amit Display cell. The cell entry reads No to indicate amitriptyline is not displayed. Examine the display of the remaining molecules and the color map.
From the Molecule Management Table, select the imip Display cell. Imipramine is removed from the viewing area. Only quinupramine is still displayed. Examine the structure and map to determine what portions of the molecule extends beyond the map boundaries.
From the Molecule Management Table, select the quinpr Display cell. Quinupramine is removed from the viewing area.
Select the amit Display cell again. The display of amitriptyline is restored. Examine it for portions of the molecule that extend beyond the map boundaries.
From the Molecule Management Table, select both the quinpr and imip Display cells. All structures are restored to the viewing area.
From the Object Management Table, select the heading cell for the Delete column. All objects are removed from the viewing area and from the table. The table is cleared.
From the Molecular Similarity palette, select Exit Molecular Similarity. The Molecular Similarity palette is removed from the screen and the Modeling palette is redisplayed.
6. Reject the changes made during the fitting procedures.
From the Modeling palette, select Reject Changes. The changes made to quinpr.msf, imip.msf, and amit.msf are not saved. The coordinates saved after minimization of quinpr.msf, imip.msf, and amit.msf are redisplayed in the viewing area.
Display the File menu and select Close. A dialog box is displayed with options for what file to close.
Select the Close All button between the two scrolling file lists. Amit.msf, quinpr.msf, and imip.msf are moved to the Close scrolling list of files.
Select the OK button. The dialog box is removed from the screen and the structures are removed from the viewing area.
Multiple conformers can be studied in the Molecular Similarity application using MSF bundles. An MSF bundle is a collection of MSFs which are related by the application through their names. All MSFs in a bundle are assumed to have the same covalent structure. Normally, the Molecular Similarity application works through a collection of MSFs, considering each MSF as a separate molecule. MSF bundles are defined to efficiently compare sets of multiple conformers as units.
Use MSF Bundles alters the way atom matching and volume calculations operate in Molecular Similarity. Normally, atom matching is specified for all active MSFs. When Use MSF Bundles is selected, matched atoms are only specified for the first MSF in each bundle.
For volume calculations, the normal procedure is to calculate an intersection map among all active MSFs. With Use MSF Bundles, volume maps are calculated for each MSF, then maps are combined by taking the union of the structures within each bundle. The resulting union maps are used to calculate the intersection between bundles.
Use the following exercise to become familiar with the use of MSF Bundles in volume calculations. This exercise uses the Conformational Search and Analysis applications. If you are not familiar with these, see chapters 3 through 6.
1. Generate six conformations of amitriptyline.
From the File menu, select Open. A File Librarian dialog box opens. Select amit.msf from the scrolling list, Replace, and the OK button.
Display the Applications menu, and select Conformational Search.
From the Conformational Search palette, select Torsions. The Torsions palette opens.
From the Torsions palette, select Define All Torsions.
From the Conformational Search palette, select Setup Search, then select Random Sampling. The Random Sampling Setup dialog box is displayed.
From the Conformational Search palette, select Do Search.
A File Librarian is displayed.
Select the New button. The conformational search begins. When it is completed, the Message Line reads:
Six different conformations of amit.msf are created.
From the Conformational Search palette, select Analysis. The Analysis palette is displayed.
From the Analysis palette, select Trace.
The Select Property for Plotting dialog box is displayed.
Select the OK button. A plot of CHARMm potential energy as a function of conformation number is displayed. A series of menus (File, Edit, Display, Trace Tools) are aligned across the top of the Trace Plot window.
Display the Trace Tools menu. Select the Select Window function. All six conformations plotted in the display are selected.
Select the File menu in the Plotting window. Select Quit. The Plotting window is cleared.
Display the Analysis palette. Select File Options/Filters. A dialog box is displayed.
Select the OK button. The Select the Output File Type dialog box is displayed.
Select the OK button. Six MSF files are created with names the same as the tags for each conformation (amit.msf:1 through amit.msf:6).
Display the Analysis palette, and select Exit Analysis. The Plot and Analysis palettes are cleared.
From the Conformational Search palette, select Exit Conformational Search. The Conformational Search palette is cleared.
From the Modeling palette, select Reject Changes to reject the current conformation.
2. Generate and store six imipramine conformations.
Open imip.msf and replace the amit.msf conformation displayed in the viewing area.
Repeat the search and analysis procedure just completed for amit.msf. Substitute imip for amit where appropriate.
3. Start Molecular Similarity Calculations.
From the File menu, select Open. A File Librarian dialog box is displayed.
Open all six amitriptyline and six imipramine MSFs by selecting each file consecutively. The files are named amit^1.msf through amit^6.msf and imip^1 through imip^6.msf.
Select the Replace button and then select the OK button. All 12 conformations are displayed, stacked on top of one another, in the viewing area.
Display the Applications menu. Select Molecular Similarity. The Molecular Similarity palette is displayed.
Select Use MSF Bundles and Match Atoms. The Match Atoms palette is displayed.
Select Pick Equivalent Atoms. A single conformation (amit^1) of the target molecule amitriptyline is displayed.
Pick the 12 carbon atoms that are to be matched with atoms in the imipramine structure.
From the Match Atoms palette, select End Atom Picking.
Both amitriptyline and imipramine are displayed in the viewing area. The match atoms in amitriptyline are accentuated by colored circles with + marks inside.
Select the corresponding match atoms on imipramine in the same order the match atoms were chosen for amitriptyline. A large cursor is present on the amitriptyline atom that needs a corresponding imipramine match.
When all match atoms have been selected, the atom matches for all 12 conformations are automatically defined.
From the Match Atoms palette, select Save Atom Matches. A File Librarian dialog box appears, requesting a name for the .mch file.
From the Match Atoms palette, select Exit Match Atoms.
From the Molecule Similarity palette, select Rigid Body Fit to Target. A fit is performed to fit all molecules to the target molecule.
From the Dial Emulator window, select the Reset View dial. The structures are realigned in the window for better viewing.
From the Molecule Similarity palette, select Calculate Volumes.
The Set Grid Resolution and Size dialog box is displayed.
An intersection volume map between union maps is calculated and displayed. For MSF bundles only this intersection map is displayed. The numerical volume is reported in the Volume Maps column of the Object Management Table and in the textport.
From the Molecular Similarity palette, deselect Use MSF Bundles.
Select Calculate Volumes. The Set Grid Resolution and Size dialog box is displayed.
A volume for each MSF is calculated and overall intersection and union maps are calculated and displayed, along with a density map. The reported volume calculated without MSF bundles is lower than the calculated volume for the MSF Bundles.
Because the intersection map is now the intersection among all the individual MSFs, the result is a lower intersection volume than that calculated between union volumes of each MSF bundle.
The Molecular Similarity application provides a visual comparison of two or more molecular structures by overlaying these structures in the viewing area. Comparisons can be made between different structures, different conformations of the same structure, and different parts of the same structure.
The only requirement is that each structure must be contained in a unique MSF. The application does not permit use of different generations of the same MSF.
The procedure used in Molecular Similarity to compare structures is divided into four basic steps:
1. Load the structures to be compared.
2. Define atom equivalences in these structures, that is, establish atom matches.
3. Perform a fitting operation.
Each structure is identified by the name of its corresponding MSF. One structure is identified as the target or fixed structure. All other structures are working or moving structures. Although only one structure can be designated as the target at a given time, the target definition can be changed from within the application.
The Molecular Similarity application makes no assumptions about the equivalency of atoms on different structures. Equivalency is defined through user input. When atom equivalencies are defined, they can be used for any fitting operation.
Fitting operations employ either a rigid or flexible method. If a rigid method is used, working structures are translated and rotated to obtain an optimum fit with the target structure. If a flexible fitting method is used, conformations of the working structures are changed to obtain an optimum fit.
The Molecular Similarity palette provides selections to rotate and translate selected structures, control the display of structures, change target structure definitions, and compare molecules. Other palette selections are used to manage the files that are generated during fitting and comparison operations.
The Molecule Management Table is used to change the activity and display of structures.
The Match Atoms procedure links equivalent or near equivalent atoms on one or more working molecules to the corresponding atom in the target structure. Atom equivalences are always defined in pairs. When you use multiple working structures, equivalent atoms are individually paired with the target atom, not collectively grouped with each other. With matches established, relationships between target and working atoms are defined, permitting sophisticated structural comparisons.
With two structure involved in a matching procedure, there are two methods available to establish atom matches:
When more than two structures are involved in atom matching, a modification of the second matching method is used.
In addition to the visual inspection of overlaid structures, it is also possible to calculate the van der Waals volume of structures and display this information in a variety of volume maps. Three types of volume maps can be displayed: union volume, intersection volume, and volume density.
Normally, the Molecular Similarity application works through a collection of MSFs, considering each MSF as a separate molecule. Alternatively, multiple conformers can be studied in the Molecular Similarity application using MSF Bundles. An MSF bundle is a collection of MSFs that are related by the application through their names. All MSFs in a bundle are assumed to have the same covalent structure. MSF bundles are defined to efficiently compare sets of multiple conformers as units.