
This utility is a graphical interface to a protein database search program. The database query is specified by entering the data in dialog boxes.The protein fragments which satisfy the search query are read into QUANTA and displayed.
The database information is derived for all the known protein structures from the Brookhaven Protein Databank and stored in the file $HYD_LIB/database.dat. The stored structural information is primary residue-based and includes some information on atoms and secondary structure. Searches to this database can address many common modeling problems. However, facilities for statistical analysis are not provided at this time.
IUPAC-IUB Commission on Biochemical Nomenclature.
Biochemistry, 9, 3471-3479 (1970).
C.M. Wilmott and J.M. Thornton J. Mol. Biol., 203, 222-223 (1988).
W. Kabsch and C. Sanders Biopolymers, 22, 2577-2647 (1983).
The protein structural information has been extracted from the current Brookhaven Protein Databank and stored in a single file usually called $HYD_LIB/database.dat.
For each protein the stored information consists of:
For each residue the stored information consists of:
Sidechain torsion angles and centers are described further in Appendix H.
You can append additional proteins to this file or create your own separate database file as described in Appendix C.
A program external to QUANTA, called SEARCH, is used to search the structural database file. This program can be run stand-alone using a control file created r within QUANTA or edited manually. The format of the control file is described in Appendix G.
Four types of information can be used to define a structure database query:
The search can be limited to those proteins with a resolution better than some specified value or using a keyword such as "`hemoglobin" in their description. However, by default all proteins are searched.
A template is a fragment of consecutive residues. Each residue may be defined by residue type (or some wildcard group of residue types such as "hydrophobic", by secondary structure type, by main chain or side chain torsions or by intra-template Ca-Ca distance. Each residue in the template may be defined as much or as little as required.
A constraint is a limiting relationship between two templates. It specifies that the two templates must be a certain distance apart or a certain number of residues apart in the sequence. For example:
If two or more templates are defined, they must be linked by constraints, otherwise they are completely independent and should be treated as separate database queries.
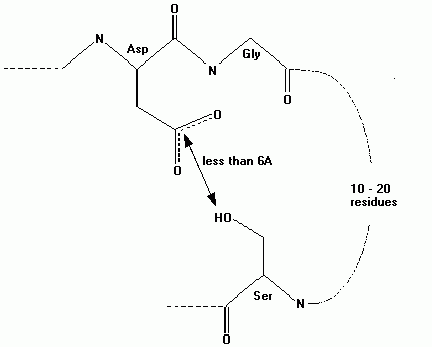
For example, to find all instances of the two residues asp and gly where the asp residue is near a ser residue that is between 10 and 20 residues ahead of the asp residue in the sequence, then the form of the query is:
Template 1: A two residue fragment with sequence asp-gly
Constraint 1: The side chain of the first residue in template 1 (asp) is between 3 Å and 6 Å away from the sidechain of the first residue in template 2 (ser).
Constraint 2: The first residue in template 2 (ser) is between 10 and 20 residues ahead of the first residue in template 1 (asp) in the protein sequence.
A database search usually finds a number of hit fragments that satisfy the query, These fragments, from different proteins, are usually superposed for easy comparison and then displayed within QUANTA.
The user can specify how tightly the search criteria must be adhered to by changing the search tolerance. For example, if you specify a required main chain or side chain torsion, then, by default, all structures within 30° of the required value will be retrieved. After you enter the required information using the Define Search tool, the control file for the search program is generated, and the search runs automatically.
The search program produces two output files: a log file that lists all hits, and a selection file. The selection file is in standard selection format, and is used by QUANTA to read in the required fragments taken from a library of MSF files. This library is a directory containing MSFs, and is pointed to by the environment variable $MSF_LIB. Each file should correspond to an equivalently named Brookhaven PDB file.
While reading in the fragments, it is optional to read in additional neighboring residues in order to display the environment of the fragment of interest. Fragments from different molecules are usually superposed so they can be compared more readily. The fragments taken from multiple MSFs are written to one MSF (along with some additional information on the database search).
The database browse tools allow rapid display and comparison of the hit fragments from one database MSF (i.e. an MSF containing fragments of multiple proteins which were found in the database search). They are the same as the browse tools found in the Fragment Database palette from the Model Backbone utility.
If more than one database MSF is selected, you are prompted to choose one to browse. When the search results are read in the MSF created automatically becomes the browse MSF. The jobname of the database search of the current browse MSF is printed at the bottom right of the screen. Each group of residues that constitute one hit are in a single segment, and the segment names that identify the fragments are displayed on the right of the screen. The segments are named with a three-letter code for the PDB file that they came from, and a single-letter code to differentiate the hits from the same PDB file.
This tool opens a series of dialog boxes for conducting a search. The first dialog is the Define Protein Structure Database Query.
A query must have at least one fragment template defined. Each additional template should be related to the existing template by a constraint. Otherwise, the templates are effectively independent and should be searched for in separate jobs. Errors in defining a template or constraint can be corrected using the List/delete option.The proteins to search and search parameters need not be entered, as the defaults are usually satisfactory. After finishing the definition of the query pick the Search button to initiate the search.
This option open the Define Template dialog box.
Enter the number of residues being searched for and a name for the template. The template name is used later for reference. If side chain torsions or intra-template distance constraints are required, toggle on the appropriate buttons. This will activate the appropriate dialog boxes that define these parameters.
This dialog box enables you to define the template residues.
It has the appropriate number of lines for the number of residues, and columns for residue type, secondary structure type, and phi, psi, and Ca pseudo-torsion angles. The residue type can be set to the wildcard ANY and boxes left blank are considered undefined.
Valid residue types are the 1 or 3 letter code for the amino acid or a wildcard. The default wildcards are defined in the file $HYD_LIB/wildcard.dat, and can be listed to the textport by toggling the option in the Define Query dialog box. To specify the wildcard type, enter the wildcard code (a maximum of six-letters) in the residue type field. The allowed wildcards can be extended or changed by editing the wildcard.dat file.
Valid secondary structure types are:
Each of these can be prefaced by NOT. The secondary structure types in the database are designated using an algorithm based on Kabsch and Sanders that is identical to the one used in QUANTA.
The valid torsion angles are in the range -180° to 180°. The torsion parameters are target values; a hit will be any residue that has torsions within the tolerance angle of that target value. The tolerance angles can be set within the Search Parameters options; the default tolerance is 30°.
If the sidechain torsions option has been selected this dialog box opens to define sidechain torsions.
As with the mainchain torsions, you enter a required target value, and all structures within the specified tolerance (default 30°) of that target value are retrieved.
If the Intra-residue distance constraint option has been selected, this dialog box opens to define intra-template Ca-Ca distance constraints.
This option is for constraints between residues in the same template. Choose the type of constraint, a target distance, and two residues are from the two scrolling lists. Note: This option is very different from the Define constraint between two templates option, which is available from the Define Protein Structure Database query dialog box.
The database contains only limited atom coordinate information: the Ca atom coordinate for each residue, and a coordinate for the side chain center. The definition of the side chain center for each amino acid type is listed in Appendix, Side Chains and Torsion Angles. This limitation means that a distance constraint between two residues can be only one of three types: a distance between two Ca atoms, a distance between a Ca atom and a side chain center, or a distance between two side chain centers. An appropriate constraint type is selected, then enter the required target distance, the default tolerance is 1.0 Å, which can be changed with the Search parameters option. Particularly when searching for distances between two side chain centers it may be necessary to search with generous tolerances on the distance criteria in order to find all required structures; structures that are not required can be rejected later after inspection.
If you click the OK button, the currently displayed definition is saved and the dialog box options are reset to the initial default values so that you can enter further constraints. Clicking the Quit and Finish buttons removes the dialog box. However, the current definition is saved only if you click the Finish button.
This option opens the Define Constraints Between Two Templates dialog box.
A constraint is a relationship between two templates. If you define more than one template, each additional template must be related to at least one of the existing templates by a constraint. Otherwise, the templates are independent and could be treated as separate queries. More than one constraint can be defined between a pair of templates. A constraint can be either a distance constraint, (e.g. the distance between a Ca atom in one template and a Ca atom in the other template), or a residue separation constraint which specifies that two templates are separated in the sequence by a given number of residues.
The database contains only limited atom coordinate information: the Ca atom coordinate for each residue, and a coordinate for the side chain center. The definition of the sidechain center for each amino acid type is listed in Appendix H. This limitation means that a distance constraint between two residues can be only one of three types: a distance between two Ca atoms, a distance between a Ca atom and a side chain center, or a distance between two side chain centers. An appropriate constraint type is selected, then enter the required target distance, the default tolerance is 1.0 Å, which can be changed with the Search parameters option. Particularly when searching for distances between two side chain centers it may be necessary to search with generous tolerances on the distance criteria in order to find all required structures; structures that are not required can be rejected later after inspection.
The obvious form of residue separation constraint is to specify a variable number of residues between two fixed patterns of residues. For example, to find a sequence pattern G-G-(2,5)X-G- two consecutive gly residues followed by between two and five residues before another gly residue- the first template is two gly residues and the second template is one gly residue. The constraint is that the first residue of the first template and the residue of the second template are between four and seven residues apart.
It also is often necessary to set an exclusion range of residues. For example, in studying side chain-side chain interactions you may require that the interactions be between residues remote in sequence. Two templates can be defined each containing one residue of the amino acid type of interest, and set an exclusion range between the two templates. If this range is set between -5 and +5 residues, then the two residues must be at least five residues apart in the sequence.
An instance where it is necessary, but not obvious, to set a constraint is if two identical templates are defined. For example, in searching for two interacting histidine residues, you would define two templates; one for each histidine residue. In this case you also must specify an exclusion range between the two templates of at least -1 to 1 to ensure that any single histidine residue does not satisfy both templates.
In other instances, it may be desirable that one residue in a protein is simultaneously in two templates. Take, for example, search for a structure with two a-helices which are connected by a loop of between 6 and 12 non-helical residues. To do this you would define two templates and set a residue separation constraint between them. The first template is 12 residues long with the first six residues specified to be helix, and the second six specified as not helix. The second template is 12 residues long, with the first six residues not helix and the second six helix. The constraint is that the fist residue of the first template, and the first residue of the second template are between 6 and 12 residues apart.
This could be illustrated by the two extreme solutions: where H = helix, and N = not-helix.
First residues in the two templates separated by 6 residues:
Template 1 H-H-H-H-H-H-N-N-N-N-N-N
Template 2: N-N-N-N-N-N-H-H-H-H-H-H
Fragment found: H-H-H-H-H-H-N-N-N-N-N-N-H-H-H-H-H-H
First residues in the two templates separated by 12 residues:
Template 1: H-H-H-H-H-H-N-N-N-N-N-N
Template 2: N-N-N-N-N-N-H-H-H-H-H-H
Fragment found: H-H-H-H-H-H-N-N-N-N-N-N-N-N-N-N-N-N-H-H-H-H-H-H
In the case which the first residues area separated by 6 residues, some residues in the hit structure are in both templates.
This option opens the Specify Proteins to Search dialog box used to specify proteins for searching. It is the same dialog used for Protein Information.
By default, any query will search the entire database until it has found the required number of hits. It is possible to limit the search to particular proteins and it is often desirable to limit the search to structures of higher resolution.
This options opens the Database Search Parameters dialog box.
In defining a search template, you enter target values for torsion angles and distances. Structures with geometry within some given tolerance of the target parameters are retrieved. The database search will end after finding some specified number of hits; the default is 50. The tolerance on the target parameters and maximum number of hits retrieved can be changed. The name of the protein database and wildcard files can also be changed. If you change these parameters, the new values remain operative through the current QUANTA session, and are saved in the protein constants file for use in subsequent sessions.
This option lists to the textport the one letter codes for secondary structure types and the amino acid wildcards currently defined in the wildcard.dat file.
This options opens the Delete Template or Constraint dialog box, which lists the current names of templates and constraints.
Click the Quit button to exit without any changes to delete a template, make a selection and click the Delete button. If a template is deleted the default is that any constraint involving that template is also deleted. This can be switched off by clicking the Delete Constraints toggle at the bottom of the dialog box.
The database search program, $HYD_EXE/search, is a separate executable which is run from within QUANTA with this option. The results are read automatically back into QUANTA. Each database search is given a job name. This name is used as a root name for the files used to transfer data between the two programs. The files are of the format: job_name.ddb. This is the query file generated by QUANTA and read by the search program. The internal format of this file is described in the Appendix, Running the Search Program. The file can be edited, and the search program run outside QUANTA.
The search program outputs two files: job_name.log is a log file that lists all the hits and reports any problems; job_name.sel, which is in the format of a QUANTA selection file, specifies which fragments of selected proteins should be read into QUANTA for display. The log file can be reviewed from within QUANTA using the Read log file option.
If you have just entered a database query and clicked the Search action button, you are prompted for a jobname. The query is written to the file job_name.ddb and the database search initiated. If the Run Search option is picked without entering a query, you are prompted to select an existing query file (.ddb). QUANTA waits while the search is run. It normally takes a couple of minutes to search the entire database with a simple structural query. Once the job is finished the results are read and displayed in the textport.
This tool opens the File Librarian to select a log file. This log file from the database search is listed to the textport. The file lists the proteins with structures that satisfy the query and the residues that correspond to the query templates. The log file also reports if the MSF file containing a hit fragment cannot be found. If you run a database query then the log file from that query is automatically listed; otherwise you are prompted to select a log file.
In order to display the results of the database query, the fragments of proteins that satisfy the query are read into QUANTA. They are then written out to a single MSF with some additional information relating to the database search in the title records of the MSF. MSFs that have been created using this utility can be displayed, and can be reviewed using the database browse facility. They are handled appropriately in other applications of the Protein Design application where the coordinates or conformations of the fragments can be copied to modeled structures.
The selection file, job_name.sel, specifies which residues of selected proteins satisfy the database query. The structural information for these fragments is read from an MSF for the protein. If the database search has found hits in protein structure for which there is no MSF file, either in the directory $MSF_LIB or in your working directory, then these hits are not included in the selection file but are reported in the log file. To display these hits an MSF will need to be created by reading in an appropriate PDB file. The MSF can be put in either the MSF library directory or in your own working directory. The database search can then be rerun so the selection file includes the relevant MSF.
If the Read Hit Fragments tool is selected without having just run a database query, you are prompted for the name of a selection, .sel, file to read. To run successfully the query file, job_name.ddb, must also be present.
This tool displays the Select Fragment to Display dialog box. It reads in and, by default, displays all the hit fragments referenced in the selection. It also enables a limited selection to be specified and read. The default setting only reads those residues corresponding to the residues of the query templates, but you can choose to read in neighboring residues.
This option, by default, reads into the initial MSF all the hit fragments. Alternatively, a range can be selected with this option. For example, selecting the range 1 to 20 would read in the first 20 hits in the selection file. In addition, specific hits can be selected from the scrolling list of all the protein MSF files in which hit fragments have been found.
This option displays two choices. They are either to select, a zone of residues, such as selecting residues contiguous with a query template, or a sphere, such as all residues close to a residue in a query template. If either of these options is picked, the appropriate dialog box to enter the selection is displayed.
This selects an extra zone of residues for each template. In selecting a zone, choose the number of residues before the first template residue, then number of residues after the last template residue. The default is zero residues, that selects no additional residues.
This selects a sphere of residues. In selecting a sphere, choose one of the template residues from the scrolling list as the center of the sphere, then enter a radius for the selection sphere. The selection uses the same criteria as NAYBR BYRES, so that all residues that have an atom within the cutoff distance of the center residue are selected. The selection is applied if you click OK or the Quit button. If you click OK the dialog box remains and another selection can be made. Click OK, for example, if you have two templates and want to select an extra sphere of residues about both templates.
This superposes hits and makes it easier to compare the hit fragments. Once the hit fragments are read in and a new MSF created by default you are automatically prompted to select the atoms to superpose. However, this option can be toggled off.
The fragments read in are all written out to one MSF. Each group of residues that constitute one hit are in a single segment. The segments are named with a three-letter code for the PDB file that they came from, and a single-letter code to differentiate hits from the same PDB file.
This tool opens the File Librarian after a search has been completed for writing the hit fragments to file. The hit fragments, by default, are written to an MSF called job_name.msf, but an alternative filename can be used.
This tool superposes fragments after you have read in hit fragments and created a new MSF. By default you are prompted to superpose the fragments. It is also possible to superpose the fragments later on using the Superpose Fragments tool.
The superposition is a conventional least squares superposition of equivalent atoms, as described in the Align and Superpose module. It depends on the context in which atoms are most appropriate to superpose. However, it is usually easier to interpret the display if you have a good superposition of one residue, by selecting just three or four atoms from that residue, rather than having a rough fit over several residues.
The dialog box to select atoms to superpose has a scrolling list of template residues. Select a residue and a text input into which you should enter the names of the atoms to superpose. If you wish to select atoms from more than one residue, click the OK button to enter the current selection, and then enter your additional selection. Click the Superpose button to initiate the superposition.
This is grayed until the fragments have been superposed. This tool saves the superpositioned fragment to the MSF. The standard saving dialog options are displayed.
This is grayed until the fragments have been superposed. This restores the previous coordinates of the fragments and the superposition is discarded.
This tool displays all hits superposed over the structure.
This tool selects the next available fragment until it has reached the last one and then it is grayed out.
This tool selects the last fragment until it has reached the first one and then it is grayed out.
This tool opens the Display Selected Fragments dialog box from which to select fragments to display.
This tool opens the Fragment Display Mode dialog box from which to select the Display options.