
This utility contains tools to calculate solvent accessibility and to calculate contact areas between two molecules or two regions of the same molecule. There are several different coloring schemes to indicate the accessibility, or contact area, of atoms, whole residues or residue side chains.
B. Lee and F. M. Richards, "The Interpretation of Protein Structures: Estimation of Static Accessibility" J. Mol. Biol. 55, 379-400 (1971).
The solvent-accessible surface area, or accessibility, of an atom is the surface area of the atom that is exposed to solvent. The residue accessibility is the sum of the accessibilities of the atoms in that residue. In studying proteins, the residue accessibility is a useful indicator to the residue's location, on the surface or in the core. One factor which should be taken into consideration in interpreting the data derived by this utility is that, because proteins are dynamic, even residues which are calculated to have very low solvent accessibility will be intermittently exposed to solvent.
In the conventional model for the accessibility calculation the solvent water molecule is represented by a sphere of radius 1.4 Å. A simple model for the calculation is of the solvent sphere being rolled over the surface of the molecule.
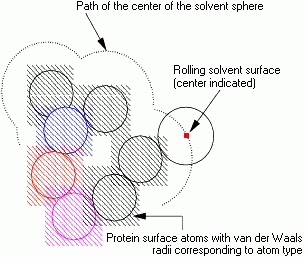
The path of the center of the solvent sphere is considered to be the accessible surface of the molecule. The distance from a protein atom center to the accessible surface is, at minimum, the van der Waals radius of the atom plus the solvent sphere radius. Because of the finite size of the solvent sphere, it cannot penetrate small interstitial spaces between atoms of the molecule surface. Hence the solvent surface appears smoother than the van der Waals surface.
Assigning a numerical value for accessibility requires a method of integration over the surface area. QUANTA uses the Lee and Richards method. It takes z-sections through the molecule, tracing a solvent accessible path around the molecule in each section. The length of the arc line is then calculated about each atom. This length is multiplied by the value of the z-spacing to give a rough estimate of area.
The z-spacing used is proportional to the sum of the solvent sphere radius and the minimum van der Waals atomic radius of the atoms in the molecule. The default proportionality constant, or z-spacing factor is 0.05.
When calculating accessibility for a protein it is necessary to exclude any associated solvent molecules and hydrogen atoms. Since accessibility calculations for a large protein can be slow, it is sometimes useful to perform the calculation for a selected set of atoms. However, a shell of neighboring atoms must be included for the context of the calculation. These neighboring atoms are responsible for occluding some surfaces of the selected atom set. When setting up a calculation for a selected set of atoms, it is probably safest and simplest to do the calculation in the context of the whole molecule.
The contact area between two sets of atoms is a measure of their surface areas in contact. The two sets of atoms might be two molecules or two regions in one protein (e.g., two neighboring a- helices).
The definition of the contact area for the first set of atoms is the area that is accessible in the absence of the second set of atoms, but occluded in the presence of the second set of atoms. A similar definition applies for the contact area of the second set of atoms. The contact area of the two sets does not have to be the same, but normally it is similar.
To find these contact areas three accessibility calculations are done:
The contact area for each atom is the difference between its accessibility in its own set and in both sets together.
To display the accessibility or contact area on the molecule there are three alternative color coding schemes: atom, residue, or residue side chain accessibility.
The maximum possible accessibility of a residue side chain obviously depends on the residue type - bigger side chains have greater surface area. The fraction of the side chain accessible can be more meaningful than the absolute value. To calculate the fraction of the side chain which is accessible you also need to know the maximum possible side chain accessibility for each amino acid type.
Estimates for the maximum possible accessibility of each amino acid side chain are listed in the file, $HYD_LIB/protein_param.dat, after the keyword ACCESS. These values were calculated for a fragment GLY-X-GLY. The backbone and the side chain of X were in extended conformation. There were no other atoms nearby to occlude the side chain, so the accessibilities of the side chain atoms were maximized.
Since the maximum accessibility of a side chain depends on the side chain conformation, this fraction is just a rough guide and can even take values greater then 1. When calculating the accessibility, there is the option to calculate the maximum possible accessibility for each side chain in its current conformation. This is done by calculating the accessibility of the side chain in the absence of any neighboring residues. This value is then used in calculating the fractional accessibility of the residue.
This option calculates the solvent accessibility of the active molecules. The Calculate Accessible Area dialog is used to specify calculation variables.
This option calculates the contact area between two sets of atoms. The Calculate Contact Area dialog is used to specify the variables for calculating the contact area. The SelectSets palette is also activated to enable you to select two sets of atoms. When you enter this palette the Select Set 1 tool is active on the Selection Utilities palette and you should select the first set of atoms using the tools on the Select Sets palette. Then pick Select Set 2 and select the second set of atoms. Pick the Finish or Quit tools from the Select Sets palette to return to the Accessibility palette.
This tool toggles coloring atoms according to their calculated accessibility.
This tool toggles coloring side chains according to their calculated accessibility.
This tool toggles coloring residues according to their calculated accessibility as a fraction of their maximum possible accessible area.
This tool toggles coloring residues that are in an unsuitable environment that is hydrophobic residues that have a high accessibility or hydrophilic residues that have a low accessibility. All other residues are colored gray.
This option displays Change Coloring Ranges dialog box. For a selected coloring mode you can change the colors used or the minimum accessibility cutoffs which define the color ranges.
This tool lists to the textport numerical information for either all atoms or all residues; those atoms or residues with zero accessibility are excluded from the list.
This tool writes the accessibility numerical information to a file with the filename MSFname_atom_access.out or MSFname_res_access.out.
This selects atom data for output. The format of the atom accessibility results is: atom number, atom name, segment ID and residue code, and accessibility of atom in square angstroms.
This selects residue accessibility for output. The format for these results is:
This excludes from the output atoms and residues with zero accessibility.
This tool saves all modifications and calculations to the active MSF as extra information with the label ACCESS and a title which you will be prompted to provide. Note that it is possible to same multiple sets of ACCESS data. The data saved with the title `Default calculated accessibility' will be used to color the molecule whenever you use the Solvent Accessibility coloring mode. It will also be used, if it exists, in some Protein Health and Profile Analysis calculations so you should only save accessibility data, calculated for the whole molecule, with this title.
This tool reads in extra information data labelled ACCESS from the MSF. If there is more than one set of data then you can select the set from the list of titles.
If you have done a calculation without saving the data to the MSF then you will be prompted to save it.