
The protein modeling tools are divided into two utilities: Model Backbone for defining mainchain conformations, and Model Side Chains for defining sidechain conformations. They are closely interdependent. Therefore, when modeling, first examine possible mainchain conformations, and then review the sidechain conformation.
Homology modeling is building a model of a protein of unknown structure based on a homologous known structure or structures. An initial model can be generated by:
Using either procedure usually results in a structure with some regions of uncertain conformation. The protein modeling tools in Protein Design are divided into two groups: one defines the main chain conformation, and the other defines sidechain conformations. While these two groups are closely interdependent, the dependency is not well understood. The best approach to take in actual modeling, therefore, is to first consider all possible main chain conformations and then consider the side chain conformations.
The Model Backbone utility has several different tools for refining a structure: regularizing, building coordinates, folding residue ranges and fragment building using the fragment database. All of these tools work specifically on the protein backbone.
All regularization in the Protein Design module uses an internal minimizer and the idealized geometry which is stored in the files
$HYD_LIB/protein_structure.gsd or the binary version of the same file $HYD_LIB/protein_structure.bgsg (see Appendix B). The regularizer tool is available in several utilities where it might to most useful: for example in the Fragment Database utility, the join between the modeled region and the rest of the structure often has poor geometry but this can be improved by regularization.
Regularization is a means of cleaning up bad geometry in a model. It is an energy minimization procedure that takes into account geometry energy terms, such as bond length, angles, and torsion. By default, in regularization van der Waals energy terms are not considered, so this method will not remove bad interatomic contacts. It is possible to set regularization to consider van der Waals interactions. While this tool will improve the appearance of the model, it should be used sparingly.
Although judgement should be used, regularization is generally chosen to model undetermined regions in the following situations:
The protein conformation can be altered by changing the main chain dihedral angles. This option provides a quick means of alternating the conformation of range of residues to a user-specified pattern.
When the backbone dihedrals are changed one end of the protein chain can remain in the same position but the moving end of the protein chain could potentially move a significant distance thorough space. There are three alternatives for determining which residues move: keeping the N-terminus fixed, keeping the C-terminus fixed, and retaining the overall average position.The large movement of one terminal is often not desired and can be avoided by using the option to break the protein chain. By default the break is made at the non-fixed end of the folded fragment but it can, optionally, be made at any point between the folding fragment and the chain terminal.
The alternative approach, to maintain the average position of the folded fragment, is done by doing a least squares superposition of the folded fragment onto the original Ca coordinates.
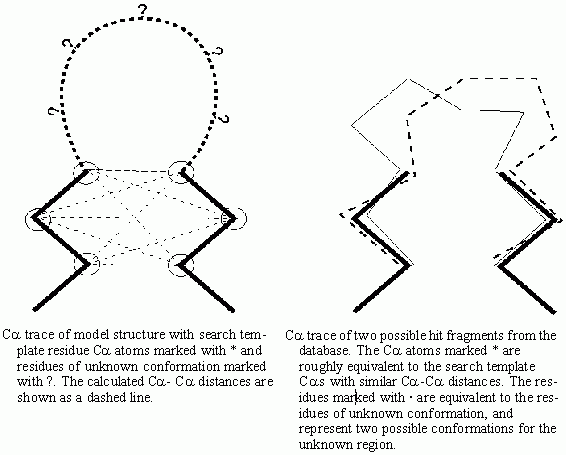
The Fragment database searching finds fragments with appropriate geometries for modeling a small region. Once a fragment is selected, its conformation is copied to the model structure.
You must select a search template of three or more residues. These
should be residues of reasonably known position either side of the
uncertain region of the model structure. For example when searching for a fragment to model a loop region between two secondary
structure elements you would pick the two terminal residues from
each of the two secondary structure elements.
The database search protocol analyzes the inter-Ca distances of the residues in the search template and searches the database for the same pattern of residues with similar inter-Ca distances.The fragments with lowest differences in inter-Ca distances are retrieved from the database and are displayed superposed over the search template. The RMS deviation for the least squares fit of the fragment over the search template is one of the parameters listed to the textport. The other listed parameter is the difference of the inter-Ca distances calculated in the database search. You can review these fragments and can select one fragment to use to model the undefined region.
Useful criteria for choosing a fragment are:
The fragment searching can also use the Bumps option. This takes retrieved fragments and fits them over the search template. The inter-atomic distances between the main chain, and Cb atoms of the fragment and the neighboring residues are calculated. Those fragments with bad close contacts are then rejected. Since this procedure reduces the number of fragments finally selected, the initial database search will retrieve extra fragments.
The fragments retrieved and displayed within QUANTA come from a library of MSF files. If this library contains compressed files then the files need to be uncompressed before reading. To do this QUANTA will create a directory TMP_MSFLIB below your current working directory and copy the uncompressed MSF files to that directory. This directory can be deleted after you have completed this work.
In the Model Backbone application, only one molecule can be active. If there is more than one active molecule when the application is entered, the first molecule is left active and the rest are set inactive. The active molecule can be changed by selecting a different molecule from the Molecule Management table.
Many of the tools in this utility are applied to a range of residues. The Pick Range tool on the Protein Utilities palette can be used to select and deselect ranges. If you pick a tool with no range selected then the Pick Range palette will be made available for you to select a range. Note that the range remains selected until you deselect it.
This option prompts you to make a selection by displaying the Pick Range palette if an active range is not selected. The Select Active Range tool on the Utilities palette can also activate the Pick Range palette. If some of the atoms within the active range are undefined, the structure is built with idealized geometry. The regularization can include interatomic interactions (so it is really an energy minimization) which can be restricted to those between atoms in the selected region or it can include interactions with the neighboring atoms. The mode of action can be changed in the Regularization Options dialog box.
Offers options on whether the interatomic interactions are taken into account in regularization.
This tool generates a structure for any undefined atoms in the structure according to the idealized geometry in the $HYD_LIB/protein_structure.gsd file but does not perform any optimization of conformation.The Pick Range palette is displayed to select a range of residues.
This option alters the protein conformation by changing the main chain dihedral angles. This option presents the Fold Protein Main Chain dialog box, from which a new fold type can be selected, and which determines the extent the structure is moved when folded.
Displays the Pick Range palette from which a range of residues can be selected. These residues need not be in the current active molecule. If the number of residues within the selected range is not equal to the number of residues in the folding fragment, then the appropriate number of residues after the first residue in the selected range will be used.
Analyze Secondary Structure and Predict Secondary Structure applications are used to assign a secondary structure type to a residue. If the residue is assigned alpha helix or strand, then it is folded to the idealized conformation for that secondary structure type. The dihedral values are taken from the main chain fold data in the file $HYD_LIB/protein_param.dat.
A dialog box with the f, y and w of all the residues in the active range is displayed. You can change the values.
Displays a scrolling list from which to make a choice from a library of idealized secondary structure types.
This library contains both repeating conformations, such as helix or strand, and structures of a finite number of residues, such as b-turn. These conformations will be applied to all residues in the active range, except for structures such as b-turns that are applied to the appropriate number of residues in the active range. The data for this library is stored in the file $HYD_LIB/protein_param.dat after the keyword FOLD. This file can be appended by the user.
Select either the Fix N-terminus end of fragment, Fix C-terminus end of fragment, or Retain average position of the fragment.
By default when the backbone torsions are changed the whole of the non-fixed terminal of the segment beyond the rotated bonds will move with the rotation. If you do not want this to happen you can opt not to carry the terminal or to carry only a limited range of residues which you will then be prompted to select.
Toggles the option to spin search the side chains to find their optimal conformation.
This option displays a palette with a set of searching and browsing tools for modeling fragments.
The inter-Ca distance matrices for a representative set of proteins is saved in the file $QNT_ROOT/dmatrix/dmfile. You can create your own versions of the file or access an alternative file by changing the file name from the Options tool. The MSF files are read from a library directory that you can set with Options tool.
The currently selected residues are indicated by a red cross on the Ca atom position and red boxes around residues on the Sequence Viewer.
Initially, all fragments are displayed superposed on the template residues. They are color coded on the structure and on the legend displayed on the right side of the screen. The legend gives the name of the protein from which the fragment is taken, its distance fit, and the RMS difference in Ca atom position when the fragment is superposed on the template.
After remodelling the protein backbone by copying coordinates from a database fragment it is advisable to remodel the side chains. The Auto Model tool on the Model Side Chains palette will do this (see Chapter 9) and, to simplify the procedure the Fragment Database utility, automatically writes a selection file, fragment_side_sel.rsd, which lists the remodelled residues.
This option list to the textport the proteins in the currently active Ca distance matrix file.
This option selects template residues by picking the first and last residue in a range.
This option templates residues by picking each individual residue.
This option deletes the last selection.
This option deletes all selections.
This option searches the fragment database by Ca distance for matches to the currently selected residues.
If this option is active, any database search is followed by Bumps checking before the optimal retrieved fragments are displayed.
This option displays all the fragments.
This option displays the next fragment on the list and removes all others from the viewing area.
This option displays the previous fragment on the list and removes all others from the viewing area.
This option opens the Display Selected Fragments dialog box with all the fragments listed, allowing you to select one or more for display.
When only one fragment is displayed this lists the residue name and ID of each residue in the fragment, and the corresponding residue in the active molecule.
This option is grayed unless only one fragment is displayed. It then copies the coordinates of the fragment onto the corresponding residues of the active molecule.
This option clears all fragments from the display
A short range of residues, by default two residues, either side of the joins between the inserted fragment and the rest of the structure are regularized to correct any poor bond lengths and angles. The range of residues which can move in the regularization can be changed under the Options tool.
This tool opens the Fragment Modeling Options dialog box which allows you to change the number of fragments displayed after a search, how fragments are displayed and the dmfile used.
This tool restores the atom coordinates to the previous state, undoing the last modeling operation.
This tool exits from the Model Backbone palette with any changes made retained in memory.