This utility superposes structures on the basis of their overall folding rather than requiring identifying homologous residues. Using this utility, protein structures with similar folding motifs, but possibly little other obvious homology, can be superposed.
A folding motif can be either a whole protein or a specific area of a protein. It is defined in terms of the a-helix and b-strand secondary structure elements, and the inter-element geometry, such as distances and angles.
E. M. Mitchell, P. J. Artymiuk, D. W. Rice; P. Willett, "Use of techniques derived from graph theory to compare secondary structure motifs in proteins," J. Mol. Biol. 212 151-166 (1989).
A. T. Brint; P. Willett, "Pharmacocophoric pattern matching in files of 3D chemical structures: comparison of geometric searching algorithms," J. Mol. Graph 5 49-560 (1987).
G. D. Mulligan; D. G. Corneil, "Correction to Bierstone's Algorithm for Generating Cliques," J. ACM 19 244-247 (1972).
C. A. Orengo, N. P. Brown; W.R. Taylor, "Fast Structure Alignment for Protein Databank Searching," Proteins 14 139-167 (1992).
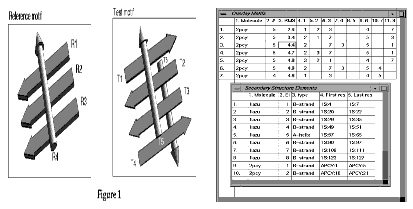
A simple example of the problem of superposing two similar, but not identical, structure motifs is shown in Figure 1.
The reference motif has a set of parallel b strands with an a-helix lying across the strands. The test motif includes a b-sheet of four strands, with the two central strands parallel and two helices, one on either side of the sheet. No consideration is made of the connectivity between the secondary structure elements.
The secondary structures are represented simply as vectors. They are labelled R1 to R4 for the reference structure and T1 to T6 for the test structure. In this example, there is no combination of elements in the test motif that reasonably matches all the elements in the reference motif. There are four possible ways in which three of the four elements in the reference motif could be matched to elements in the test motif.
For this example, it is possible to determine all possible matches by eye. However, for more complex examples, an efficient algorithm to test all possible combinations is needed.
In the automated matching algorithm each structure is analyzed separately. The geometric relationship (i.e., distances and angles) between all pairs of secondary structure elements are calculated. These geometric relationships are then used to determine whether a pair of elements in one structure might be equivalent to a pair of elements in the other structure. If the differences in distances and angles are not to great then the two pairs of elements might be equivalent.
|
R1
|
T2
|
-
|
T3
|
-
|
|
R2
|
T3
|
T2
|
T2
|
T3
|
|
R3
|
-
|
T3
|
-
|
T2
|
|
R4
|
T5
|
T5
|
T6
|
T6
|
For the example in the figure the distance and angle between the two elements of the reference structure, R2 and R4, are similar to the distance and angle between the elements of the test structure, T2 and T5. However, they are dissimilar to the distance and angle between elements T1 and T5, since the strand T1 is running in the opposite direction.
The result of comparing all possible pairwise combinations of elements is recorded in a correspondence matrix which records either true or false for whether pairs of elements might be equivalent. A graph theory algorithm is used to analyze the correspondence matrix to find combinations of elements in the reference structure which will match the maximum number of elements in the test structure. Frequently there are several possible combinations of elements which give the same number of matches overall.
Given a set of possible matches between secondary structure elements the two structures can be superposed. This is done by applying a standard least squares superposition algorithm to the endpoints of the axial vectors of the matching elements. Combinations of matches which lead to poorly superposed elements (i.e., with a poor rms difference in the endpoint coordinates) can be removed from the list of matches.
The axial vector of a secondary structure element is defined as the principle moment of the Ca atom co-ordinates. The endpoints of a vector are the projection of the terminal Ca atoms onto the vector. The relationships between pairs of axial vectors are defined as:
This is the minimum distance between the two axial vectors. When the closest point on one vector from another, is the vector endpoint, then the minimum distance is to that endpoint, rather than to any point on the line extended beyond the vector.
This is the average of the distance between all the Ca atoms in one secondary structure element and all the Ca atoms in the other element.
This angle is derived from the inverse cosine of the scalar product of normalized vectors.
This angle uses the definition given by Orengo1 for representing the relative orientation of two axial vectors as two angles.
If the protein structures have been analyzed into domains then to satisfy "same domain" criteria pairs of secondary structure elements should have the same relationship "in same domain" or "not in same domain".
The fold matching algorithm does not inherently require that matched secondary structure elements are in the same order along the protein chain but this requirement can be set.
Secondary structure elements are represented by axial vectors. The Secondary Structure tool from the Protein Utilities palette will toggle the display of secondary structure vectors.In this utility it is recommended that the secondary structure vectors are displayed, but the molecule visibility be switched off.
To superpose only a limited fragment of a protein some secondary structure elements can be made inactive. The activity is toggled using the Change Activity tools on the palette. Secondary structure vectors that are active are represented by vectors with fat lines; and those that are inactive are represented by vectors with thin lines.
If structures have been matched with the criterion that elements have the same connectivity, it is reasonable to attempt to find an optimal alignment of the protein sequence. The structures are superposed, and an alignment is performed to minimize the distance between Ca atoms in aligned residues.
This tools on this palette can be grouped interfere main functions: selecting the active secondary structure elements, matching the secondary structure, reviewing the matches and superposing and aligning structures based on one selected match.
All secondary structure elements in a molecule are used by default. However, elements can be made inactive or unselected using one of the selection tools.
When Change Active is picked the Pick Element, Pick Element Range, and Pick Domain tools are ungrayed. Only one of these tools can be used at a time.
This option is used to toggle on and off the activity of a single secondary structure. The activity is toggled by picking an atom or residue. This is done either on the structure or sequence table, of a secondary structure element.
This option is used to select the activity of any two secondary structure elements. Either two atoms in the molecule or two residues in the sequence table. All elements within the selected range of the two elements are toggled on or off.
If domain analysis has been performed on the molecule, this option selects one of the assigned domains. The activity is toggled by picking an atom or residue from the sequence table of a selected domain. If domains are unassigned, any pick selects a complete segment.
When this tool is selected, all possible overlays for the two active molecules are calculated and the resulting vectors displayed. In the legend area, each overlay is numbered and listed, along with RMS difference after superposing the secondary structure vectors. Two tables are also displayed that show information about the motif overlays and secondary structures.
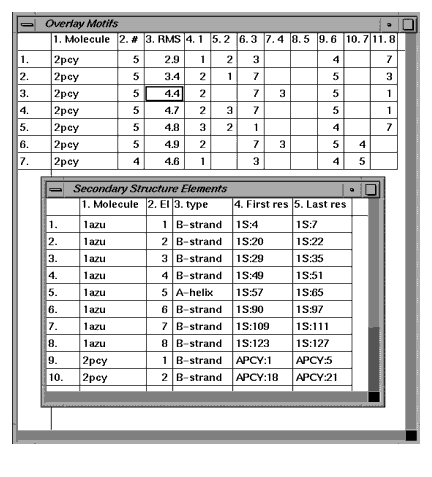
These tables display information about the calculated motifs and secondary structures for each molecule.
The Overlay Motifs table displays numerical information on each of the possible overlays. Columns are, from left to right: the overlay number, the second molecule name, the number of elements matched, the RMS difference of the superposition. All subsequent columns identify elements that are matched. For example, column 6.3, row three has the number seven. This indicates that the third element in 2pcy was matched with the seventh element in 1azu.
The secondary structure elements table displays information on each secondary type in both active molecule. Columns are, from left to right: molecule name, element number, secondary structure type, the ID of the first residue in the secondary structure, and the ID of the last residue in the secondary structure.
This tool displays the Motif Superposition dialog box that contains variables used in matching secondary structure elements and the match cut-offs. These variables set the minimum criteria for structures to be considered matched.
Only overlays which match a minimum number of secondary structure elements will be reported in the Motif Table and displayed. There are two alternative means to define the minimum.
This is the root mean square (rms) difference in the coordinates of the ends of the axial vectors after superposition. This can be used as a test for the similarity of the position, orientation, and length of the vectors and if a match results in a poor overlay then it will be removed from the list of matches.
.The individual matched elements should satisfy the following criteria:
These are criteria fora pair of secondary structure elements in one structure to be considered similar to a pair of secondary structure elements in the other structure. Most of these criteria relate to the distances and angles between the pairs of elements.
This tool is inactive until the Overlay Motif option is selected and calculated. It displays all of the calculated overlays in the viewing area and lists them with their rms values in the legend and in the textport.
This tools is inactive until the Overlay Motifs tool is used and there is more than one match. This tool steps forward displaying each overlay.
This tool is inactive until Overlay Motifs tool is used and there is more than one match. This tool steps backward displaying each overlay.
This tool is inactive until Overlay Motif tool is used. It presents you with a list from which to select one or more overlays to be displayed.
This tool is inactive unless Overlay Motifs tool is used. It removes the display of overlays and masks the browse tools.
This is inactive unless one match is selected by the browse tools. Superpose the molecule co-ordinates on the basis of the displayed match. If the molecules are invisible then make them visible.
This tool rereads the MSF and restores the atomic co-ordinates.
This tool saves the current atomic co-ordinates to the MSF.
When one overlay is selected this tool aligns the sequence based on minimizing the distance between Ca atoms. The result of this alignment probably is only meaningful if structures are matched with elements in the same order.
This discards the current alignment.
When sequences are aligned, this tool indicates which pairs of aligned residues are close by placing yellow bars on the sequence viewer. This is similar to the Match Residues option on the Align and Superpose palette. The cutoff criterion for close residues is, by default, 2.5 Å.
Exit the Superpose Folding Motif palette. If molecules have been superposed but not saved, you are prompted to save coordinates to the MSF.
The following exercise demonstrates how to us the Superpose Motif palette. The active structures used in this example are 1azu and 2pcy.
1. From the Molecule table, toggle the activity on and visibility off for structures 1azu and 2pcy.
2. From the Protein Utilities Menu, toggle on the tools Secondary Structure and Legend. Next, select the option Molecule Colors, and from the Molecule Colors dialog box.
Color Mode
Secondary Structure
Color non-carbon atoms by element type color
Select Atoms to Display
Alpha Carbon atom trace
Click OK and the display and legend are updated, reflecting the selected changes.
3. From the Protein Design Menu, select the utility Superpose Folding Motif.
4. From the Superpose Folding palette, select the tool:
The overlays are calculated, using the default Motif options, and displayed in the viewing area. The motif tables, Overlay Motifs and Secondary Structures Elements, are displayed and the browse tools are unmasked and activated. The legend list all the overlays along with their RMS value.
The first overlay is displayed in the viewing area and legend. Click on the tool to step forward through the overlays, or, to step backwards, use the tool;
6. View the Overlay Motif Table. The calculations resulted in seven possible overlays, and these are listed in order of increasing rms value.
7. Select the tool Select Overlay(s)...
The Display Selected Fragments dialog box is displayed. Pick from the scrolling list overlay 1.
and the tools Superpose Molecule and Align Sequence are automatically selected.
The structure 2pcy is superposed on the 1azu molecule; the 2pcy sequence is aligned to 1azu to minimize Ca-Ca distances; and matches between the two molecules are calculated and reported in the textport.
The molecule 2pcy is reread into the work area, and the coordinates are restored to the saved version of the MSF.
The Superpose Folding Motif utility closes.